Detect QIAseq Human Indentity SNPs and Microhaplotypes
The workflow Detect QIAseq Human Identity SNPs and Microhaplotypes (beta) (figure 7.1) can both be used to analyse data from SNP and microhaplotype panels. The default parameter values are set for mixed sample sensitivity that allows detection of a human genome present in 10% of the reads.
Note:
- In order to detect alleles present in 5% of the reads, the read coverage of the locus must be at least 200.
- There is not yet any special homopolymer handling, so length variation artifacts must be considered when analysing loci around longer homopolymers.
The Detect QIAseq Human Identity SNPs and Microhaplotypes (beta) workflow can be found here:
Toolbox | Template Workflows | Biomedical Workflows (![]() ) | Detect QIAseq Human Identity SNPs and Microhaplotypes (beta) (
) | Detect QIAseq Human Identity SNPs and Microhaplotypes (beta) (![]() )
)
Double-click on the workflow to run the analysis.
The following parameters can be adjusted when running the workflow:
- Minimum group size
- Increase to disregard UMI reads with low number of supporting reads.
- Minimum average quality score
- Increase to disregard UMI reads with low quality.
- Maximum phasing distance
- The default value is set for microhaplotypes. It can be set to 1 when analysing SNP panels where marker loci are close to each other, and phasing is not of interest. This may for example be useful when analysing data from the Ancestry and VISAGE SNP panels. Reducing this value may decrease processing time.
- Minimum count (call)
- Read support threshold for called alleles. Alleles with fewer supporting reads can be inspected by enabling 'Show filtered' in the side panels.
- Minimum allele fraction (call)
- Allele fraction threshold for called alleles. Increase this parameter (e.g. to 0.15) to remove noise when the sample only contains the genome of a single individual.
- Minimum haplotype quality (call)
- Quality threshold for called haplotype alleles. Haplotypes with lower quality can be inspected by enabling 'Show filtered' in the side panels.
- Minimum coverage
- Loci with lower coverage are marked as filtered and can be inspected by enabling 'Show filtered' in the side panels.
- Minimum average base quality
- Apply a filter to an allele when UMI read bases supporting the allele have lower average quality than this threshold.
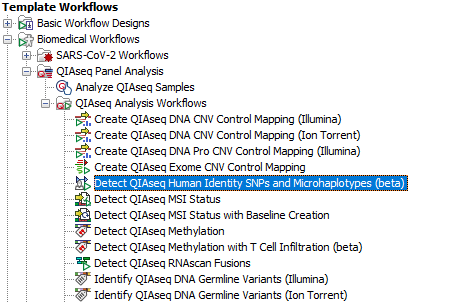
Figure 7.1: The QIAseq SNP and microhaplotype panel analysis workflow.