Upload to QCI Interpret
The Upload to QCI Interpret tool uploads a sample to QCI Interpret or QCI Interpret Translational from several types of tracks and reports.
To run Upload to QCI Interpret:
Toolbox | QIAseq Expert Tools (![]() ) | QCI Interpret Integration (
) | QCI Interpret Integration (![]() ) | Upload to QCI Interpret (
) | Upload to QCI Interpret (![]() )
)
In the tool wizard, choose which elements to upload and how to log in.
The first step defines inputs. Supported input elements are:
- Variant track(s) Add variant tracks to include variants. Choosing multiple variant tracks exports all variants in the variant tracks, for duplicates only the variant with the highest quality is exported.
- CNV track Add a CNV track to include copy number variations.
- Fusion track Add a fusion track to include fusions.
- Inversion track Add an inversion track to include inversions.
The next step is to define how the QCI Interpret sample will look in QCI:
- Sample name The name of the uploaded sample shown in QCI Interpret. Leaving it empty will use a name of a element. Use metadata if running as part of a workflow (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Configuring_input_output_elements.html)
- Subject ID Set the subject ID, leave empty, or use metadata if running as part of a workflow.
- Project Set the project, leave empty or use metadata if running as part of a workflow.
The next step defines a reference and extra values that can be added to the sample:
- Reference sequence track To generate the VCF the tool needs a reference track. Hg19 and hg38 references are supported.
- TMB report Include tumor mutation burden value and status by including a report output from the Calculate TMB Score tool in the Biomedical Genomics Analysis plugin. The algo must have been set up to detect a TMB status.
- MSI report Include microsatellite instability status by including a report output from the Detect MSI Status tool in the Biomedical Genomics Analysis plugin.
The next step defines how the uploaded VCF will be created. The Prefill VCF settings changes the VCF settings to suggested defaults depending on the data. The somatic and germline/hereditary workflows of QCI Interpret focus on different needs, with somatic being primarily focused on therapeutic, prognostic, and diagnostic actionability, while germline/hereditary is better suited for disease diagnosis/risk. Read more about VCF export settings in http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Export_in_VCF_format.html.
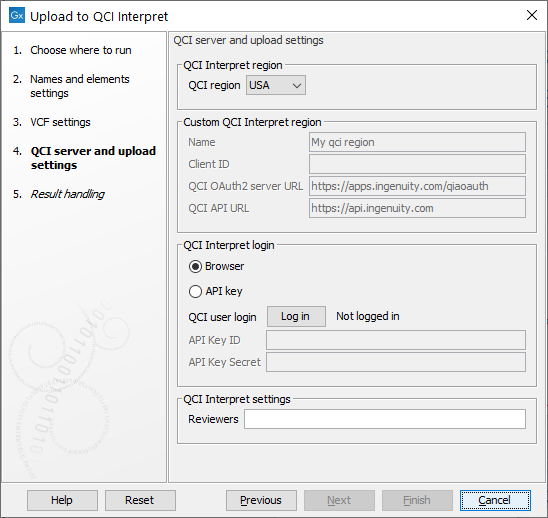
Figure 15.1: Choosing QCI region and log in.
In the final step choose your QCI Region and how to log in. See figure 15.1. To log in using a browser, choose the Browser option, then click the Log in button to open a new browser (or new tab) where you can log in, and give the workbench permission to upload samples to QCI on your behalf. To upload with API key you need an API user account, please send an email to support-license@qiagen.com and ask for an API-enabled QCI Interpret account. The license team can then send you your API Key ID and API Key Secret.
Note: Do not share workflows that contain your QCI Interpret login information. A workflow containing this tool contains your login information if the tool configuration has values set for API Key ID, or API Key Secret, or if you have logged in using a browser.
The uploaded sample can be shared with QCI users by adding a comma separated list of emails in the Reviewers setting.
Output
The output is a report that shows a summary of the data in the sample, information about the upload, and a link to the sample list in QCI Interpret.