Create Whole Genome Alignment
Create Whole Genome Alignment works by identifying seeds, i.e., short stretches of nucleotide sequence that are shared between multiple genomes but not present multiple times on the same genome. These seeds are then extended using a HOXD scoring matrix [Chiaromonte et al., 2002] until the local alignment score drops below a fixed threshold. From the initial extended seed matches, a distance matrix between the input genomes is calculated. This distance matrix is used for the subsequent pairwise processing, where the most similar genomes are processed first. Proceeding iteratively on the most similar pair of genomes, the tool will then extend and merge seed matches to create longer alignment blocks. These blocks may be present on two or more genomes, and may align to both strands of the genomes (allowing for the identification of inversions). Similar to progressiveMauve [Darling AE, 2010], we combine the HOXD substitution score with an adjustment term based on kmer frequency. This is done to avoid spurious matches to repetitive regions in the genome.
To run Create Whole Genome Alignment, go to:
Tools | Whole Genome Alignment (![]() ) | Create Whole Genome Alignment (
) | Create Whole Genome Alignment (![]() )
)
Once the tool wizard has opened (figure 3.1), choose two nucleotide sequences or nucleotide sequence lists. If the input objects are nucleotide sequence lists (chromosomes or contigs) , each sequence in the list is considered to be part of the same genome.
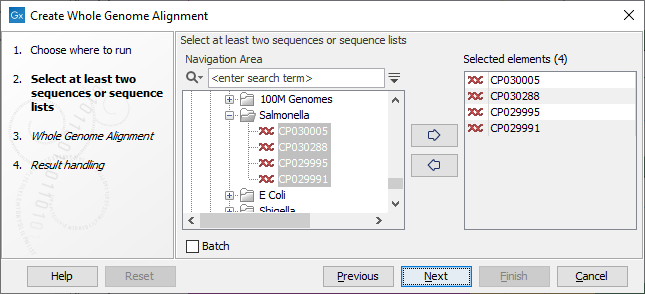
Figure 3.1: Select input for the Create Whole Genome Alignment tool.
The tool has the following alignment options (figure 3.2):
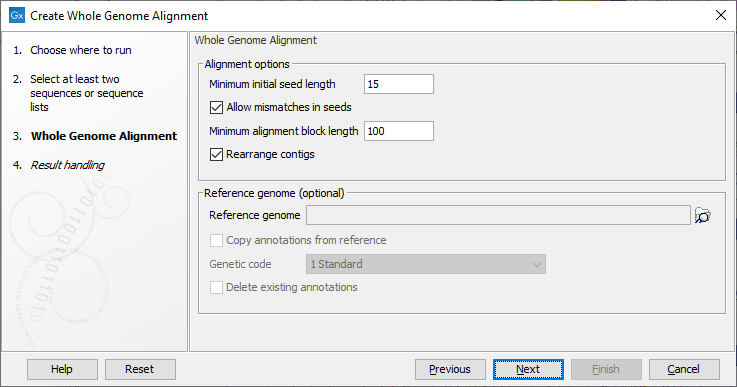
Figure 3.2: Configurable parameters for the Create Whole Genome Alignment tool.
- Minimum initial seed length The tool works by identifying seeds - short stretches that are similar - in the genomes. This option determines the number of nucleotides required for a seed to be included in the later stages of the alignment. Note about memory requirements: as a rule of thumb, the tool needs 10 bytes of memory for each nucleotide in the input genomes, plus additional memory for storing the found seeds. This means that smaller values of the "Minimum initial seed length" option will require more memory. As a consequence, the tool is not designed for large eukaryotic genomes, but for aligning bacterial size genomes.
- Allow mismatches in seeds When this option is enabled, the search for initial seeds will allow for mismatches in the seeds. This makes it possible to visualize more divergent genome pairs, but may also introduce more noise.
- Minimum alignment block length Only alignment blocks larger than this length will be output. This parameter may be used to filter away smaller and potentially erroneous alignments.
- Rearrange contigs If the 'Rearrange contigs' option is enabled, the input genomes will be rearranged to more closely match each other. The possible rearrangements are: reordering the contigs in each genome, reverse complementing contigs, and shifting of circular contigs. Rearrangements are performed in order to minimize the number of crossing lines between alignments blocks in the final whole genome alignments. Notice, that if a reference genome is specified, the rearrangements will be performed to most closely match the reference genome. The reference genome itself is not modified. If no reference genome is specified, preference will be given to the genome with the highest average contig length, though it may still undergo rearrangements.
The tool also has options for working with a reference genome (figure 3.2):
- Reference genome This option can be used to specify a reference genome. A reference genome can be used when copying annotations (see below), or to guide the rearrangements (see the Rearrange contigs option above). It will not make a difference whether the specified reference genome is included as part of the input genomes or not - in either case, only a single copy of the reference genome will appear in the whole genome alignment.
- Copy annotations from reference When enabled, annotations from alignments blocks are copied from the reference to all aligned genomes. Copied annotations will have an added 'copied_from=GENOME_NAME' qualifier. Annotations on the reference sequence may extend beyond alignments blocks, in which case the transferred annotations become partial copies: These annotations will have an additional 'partial_copy=true' qualifier. For CDS annotations, the reading frame may change. To avoid this, all 3 possible reading frames are checked and the one with the fewest stop codons is chosen. The best reading frame is annotated using a 'codon_start' tag. The 'genetic code' parameter is used when identifying stop codons. If the best reading frame still contains (non-trailing) stop codons, a qualifier 'copy_contains_stopcodons=true' is added to the annotation. Any pre-existing translation, codon_start, or frame qualifiers present on the original reference annotation, are removed on the copied annotations.
- Genetic code Used when transferring CDS annotations (see above).
- Delete existing annotations When enabled, all annotations on the input genomes will be removed.
The tool outputs a Whole Genome Alignment showing the aligned regions between the genomes (figure 3.3).
The output option Output genomes after alignment makes it possible to output the genomes, including any modifications, such as contig rearrangements and added annotations. This can also be used in workflows for automated annotations of genomes against a reference genome.
Note that the tree that is output per default in the Whole Genome Alignment view is a Neighbor-Joining tree based on the distance matrix (see the beginning of this section).
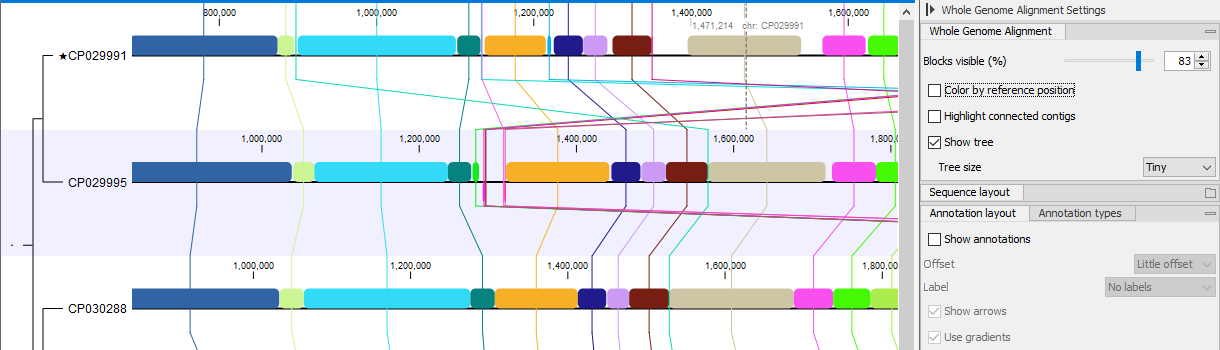
Figure 3.3: Whole Genome Alignment view. The star next to the top genome name indicates that this genome was chosen as a reference.
An alignment block (shown as a colored box) corresponds to a region of the genome that is aligned to a region on at least one other genome. The position of the box relative to the sequence indicates the strand on which the alignment was identified: above the sequence for the plus strand, below the sequence for the minus strand. When hovering the mouse over a block, the corresponding alignment blocks on the aligned genomes will be highlighted. The connected alignment blocks (which will share the same color) can be thought of as an ordinary linear multiple sequence alignment: they will not contain any internal rearrangements.
When clicking on a position on a genome, the view will automatically modify so that the aligned positions are centered on top of each other. When double clicking an alignment block, the regions covered by the connected alignment blocks will be selected.
The Whole Genome Alignment view shares most of the functionality of the ordinary sequence viewer: this includes the ability to show any annotations on the genomes (such as CDS or Gene annotations), searching for gene names (using the "Find" panel), and zooming down to the nucleotide level.
The Whole Genome Alignment view has a few special options:
- Blocks visible (%) This option makes it possible to hide the smallest alignment blocks. A value of 20 means that only the longest 20% of the alignment blocks will be shown. The default value is chosen such that 100 alignment blocks are visible.
- Color by reference position Alignment blocks are colored according to their position on the chromosomes/contigs of the reference genome. This option is only available if a reference genome is chosen. Reference genomes are indicated by star prepending the genome name (see figure 3.3).
- Highlight connected contigs When hovering an alignment block, chromosomes/contigs not part of the alignment will be dimmed.
- Show tree When the editor is opened, a tree is shown to the left of the genomes. This is the tree created by the Create Whole Genome Alignment tool using Neighbor-Joining on the distance matrix calculated from the extended seeds. This tree will provide a rough overview of the relations between the genomes: closely related genomes should be located close to each other. It is possible to change the ordering of the genomes by dragging them in the view, but this requires that the "Show tree" option in the side panel has been disabled.
Extracting multiple sequence alignments: When selecting part of a sequence in an alignment block, it is possible to use the context menu to extract the selection into an ordinary multiple sequence alignment (figure 3.4):
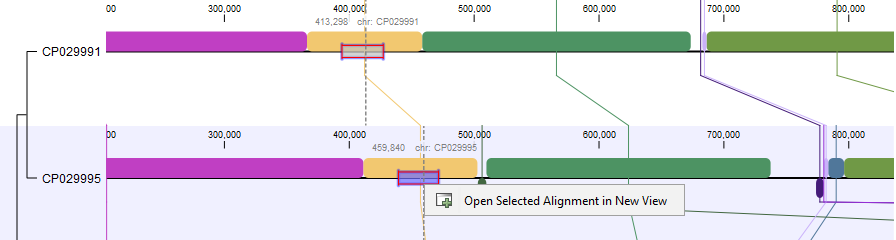
Figure 3.4: Whole genome alignment viewer.
Open as Sequence List in New View: When using the context menu on a genome (figure 3.5), it is possible to open the genome as a new sequence list, including any re-ordering, shifting, and reverse complementing done as part of the alignment.
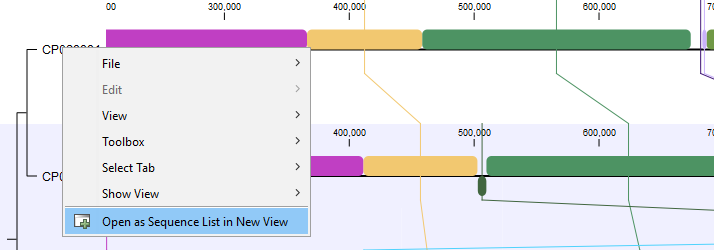
Figure 3.5: Whole genome alignment viewer.