Running the duplicate reads removal
The tool is found in the Toolbox:
Toolbox | Resequencing Analysis (![]() ) | Remove Duplicate Mapped Reads (
) | Remove Duplicate Mapped Reads (![]() )
)
This opens a dialog where you can select mapping results in read tracks (![]() ) format. Clicking Next allows you to set the threshold parameters as displayed in figure 22.35.
) format. Clicking Next allows you to set the threshold parameters as displayed in figure 22.35.
Figure 22.35: Setting the stringency for merging similar reads.
The parameter is explained in detail in Algorithm details and parameters.
Clicking Next will reveal the output options. The main output is a list of the reads that remain after the duplicates have been removed. In addition, you can get the following output:
- List of duplicate sequences
- These are the sequences that have been removed.
- Report
- This is a brief summary report with the number of reads that have been removed (see an example in figure 22.36).
Note! The Remove Duplicate Mapped Reads tool may run this before or after local realignment. The order in which these two tools are run should make little if any difference.
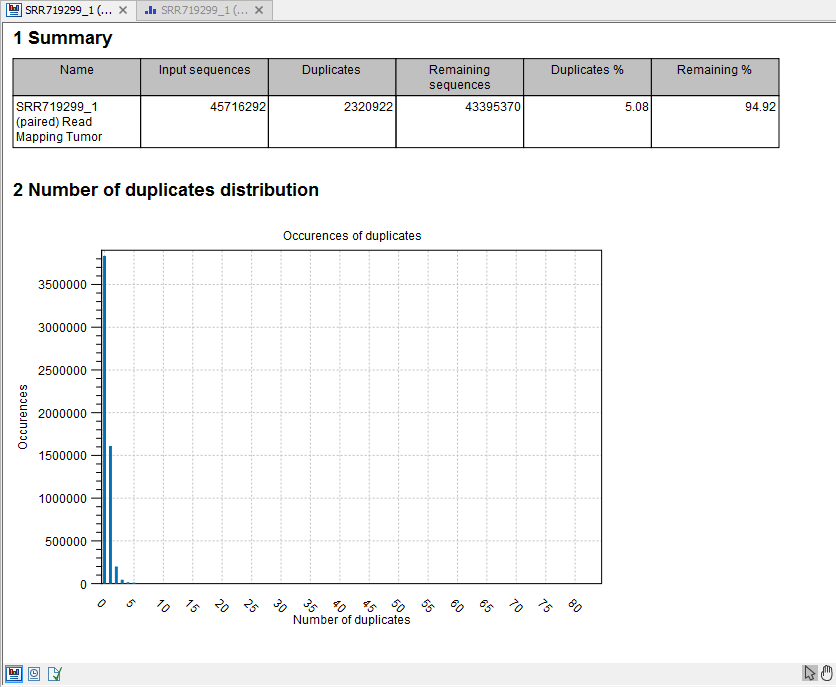
Figure 22.36: Summary statistics on the duplicate mapped reads.