Ready-to-Use Workflows descriptions and guidelines
Biomedical Genomics Workbench contains several ready-to-use workflows that support analysis of cancer data, but also analysis of hereditary diseases and other conditions that are best studied using family analysis.
Before running an application workflow, it is important to prepare the sequencing reads as explained in the following diagram (figure 12.1).
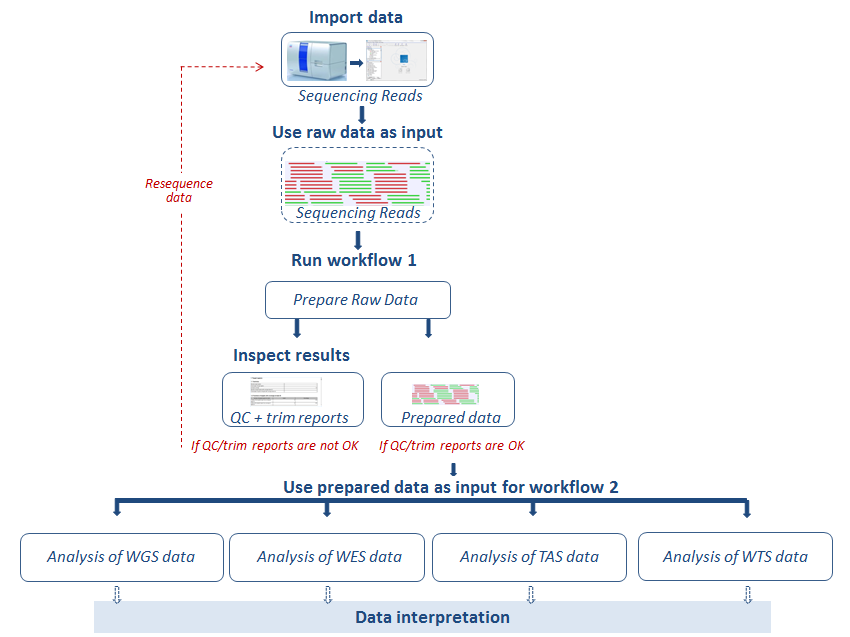
Figure 12.1: From sequencing reads to data interpretation.
The workflows are specific to the type of data used as input: Whole Genome Sequencing (WGS), Whole Exome Sequencing (WES), Targeted Amplicon Sequencing (TAS) and Whole Transcriptome Sequencing (WTS). For each of the first three categories, WGS, WES, and TAS, workflows exist that can be used for general identification and annotation of variants irrespective of disease, these workflows are found in a folder called General Analysis. In folders called Somatic Cancer, you can find workflows that are specific for cancer research. Finally, you will find a folder under each of the WGS, WES, and TAS applications, that is labeled Hereditary Disease. The workflows found in this folder can be used for studying variants that cause rare diseases or hereditary diseases (HD).
The ready-to-use workflows found under each of the first three applications have similar names (with the only difference that "WGS", "WES", or "TAS", or have been added after the name). However, some of the workflows have been tailored to the individual applications with parameter settings that have been adjusted to fit e.g. the expected differences in coverage between the different application types. We therefore recommend that you use the ready-to-use workflow that is found under the relevant application heading.
Subsections