Running tools and workflows
When CLC Single Cell Analysis Module is installed, the toolbox is populated with Single Cell Analysis specific tools and workflows. These include single cell related importers described in Data import, and single cell related exporters described in Data export. In addition, the Single Cell Analysis folder will appear in the Toolbox as shown in figure 1.1, and the Single Cell Workflows folder will appear in the Template Workflows section as shown in figure 1.2. To launch a tool or workflow either double-click it in the toolbox or use the quick launch button (figure 1.10). If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
Figure 1.10: The Launch button is situated in the top left corner of the workbench.
Running tools
All tools require an input. In many cases this will be an imported Expression Matrix (![]() ) / (
) / (![]() ), Peak Count Matrix (
), Peak Count Matrix (![]() ), or Cell Clonotypes (
), or Cell Clonotypes (![]() ), or, when starting from FASTQ, imported reads.
), or, when starting from FASTQ, imported reads.
Clicking the Import button in the top toolbar will bring up a list of supported data types, see figure 1.11. Select the appropriate importer and follow the wizard steps. For additional guidance on import settings please refer to the relevant section of Data import. Save the imported data in the navigation area. This is most easily done by choosing to save and selecting a save location. After import, the data can be found in the navigation area, ready for analysis.
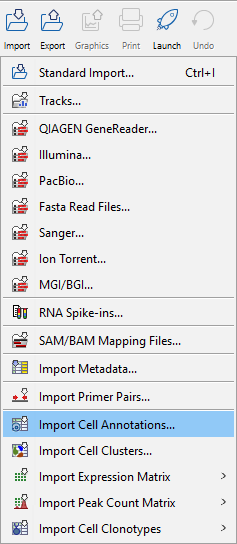
Figure 1.11: Import options with single cell related importers in the lower part. Several of the importers can be expanded to select among multiple formats.
Tools will only run correctly when meaningful input and parameters are provided. If a warning is shown, read it carefully as it will in most cases guide you to the problem.
Select the input based on the description provided in the tool. Although most tools will only allow selection of the correct data type as input, it may still be possible to provide input that does not conform to the tool's expectations. If this happens, the tool will give a warning or gray-out options that cannot be used. The default settings for each tool are suitable for most use cases, but some settings require knowledge of sample preparation or sequencing technology. Most problems arise from inappropriate settings. If a tool produces a report, make sure to inspect it, as it may provide quality control or guidance for how to change settings.
For a full description of running tools, handling results and batch options see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Running_tools_handling_results_batching.html.
Running workflows
CLC Single Cell Analysis Module provides template workflows for analyzing scRNA-Seq, scATAC-Seq, and scTCR-Seq data in multiple steps. Input can either be selected from the navigation area or imported using Select files for import. Make sure to select the correct import type.
Follow the instructions in the wizard steps and save the data by specifying a location in the navigation area. Read more about the provided workflows and how to select proper parameters in Single cell template workflows from reads and Single cell template workflows from imported data.
For a full description of how to launch workflows and use metadata see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Launching_workflows_individually_in_batches.html.