Set Up Microbial Reference Database
The Set Up Microbial Reference Database tool adds metadata to individual sequences in a sequence list so microbial isolate samples can be analyzed in the context of this metadata for typing. In general, the tool will be used to associate a user's own metadata to reference genomes included in a pathogen reference genome database (such as the one downloaded by the Download Microbial Reference Genomes tool).
The tool import pathogen information to an existing database and bundle the new metadata or overwrite the existing metadata reference by reference if this option is selected. This means that references that already have metadata and for which no new metadata is imported will keep the metadata; references with metadata for which new metadata is imported will be updated; references with no metadata will acquire new metadata.
To run the tool, go to:
Microbial Genomics Module (![]() ) | Databases (
) | Databases (![]() ) | Taxonomic Profiling | Set Up Microbial Reference Database (
) | Taxonomic Profiling | Set Up Microbial Reference Database (![]() )
)
In the first window, choose a new sequence list or an existing database (figure 15.4).
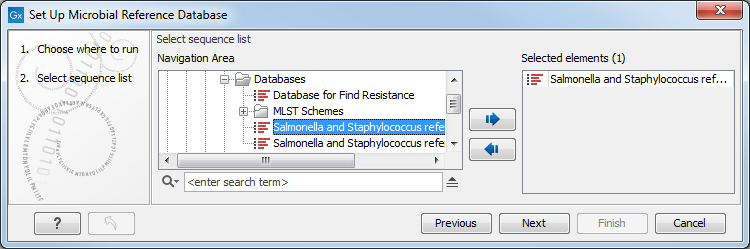
Figure 15.4: Select the existing database you want to edit.
In the second dialog (figure 15.5), select an Excel spreadsheet or a CSV file saved on your computer.
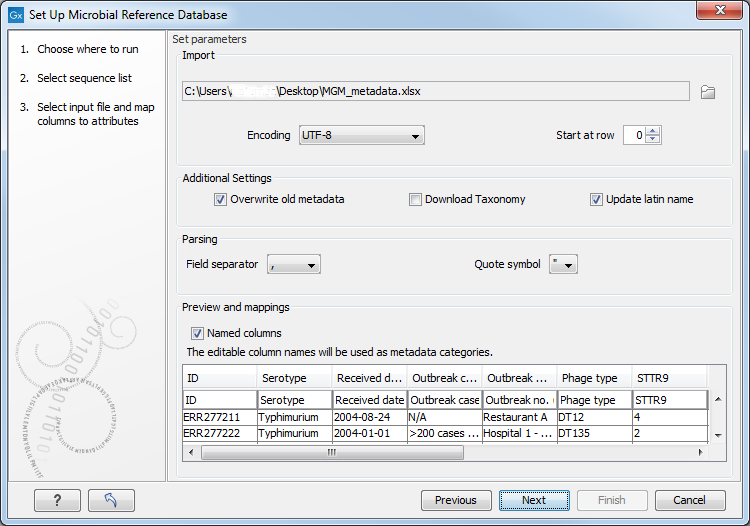
Figure 15.5: Select the file containing the metadata you wish to bundle with your existing database.
The input file must have a Name column to ensure that the link between the sequence and the metadata is successful. In addition, it is highly recommended - but optional - to have a Taxonomy column for use with other downstream tools (especially in cases where the sequence list does not have this information already). Taxonomy must be following the Qiime or common 7-level formats.
- Qiime format, such as "k_Bacteria, p_Proteobacteria, c_Gammaproteobacteria, o_Enterobacteriales, f_Enterobacteriaceae, g_Klebsiella, s_Klebsiella pneumoniae"
- The common 7-level, semi-colon separated format such as "Bacteria; Proteobacteria; Gammaproteobacteria; Enterobacteriales; Enterobacteriaceae; Klebsiella; Klebsiella pneumoniae", going from (super)kingdom to species.
You can add as many columns of metadata as you wish. The names given in the first row will be used as metadata categories. Once imported, the information from the spreadsheet (or CSV file) will fill in the table included in the lower part of the wizard window (see figure 15.3), and the headers will take the names of the first row. It is still possible to edit the first row data at this point, thereby changing the names of the metadata categories. Leaving a first row field blank means that the metadata in that column will not be imported.
The tool will output a single sequence list containing both sequence data and metadata for each sequence initially present in the input sequence list. By selecting the "Overwrite old metadata" option, the references already associated with metadata for which new metadata is imported will be updated.
In the last wizard window, the option to generate a report is selected by default. The report contains the following summary information:
- Number of sequences in the input sequence list, and in the output sequence list
- Number of sequences skipped because of duplicated names
- Sequences left unchanged and sequences for which metadata was updated
- Number of metadata columns in the input metadata table
- A list of all the metadata columns in the input metadata table
- Number of kingdoms, phyla, classes, orders, families, genera and species present in the imported database
- Number of sequences for which the taxonomy could not be interpreted. It is possible to edit the relevant entries in the sequence list table manually by right-clicking on the relevant field.