RNA-Seq normalization
Many RNA-Seq tools compare samples based on their read counts. Because sequencing depth can vary between samples, library size normalization must be performed before the samples can be compared. This section provides an overview of the normalizations used.
Library size normalization calculates a per-sample normalization factor to weight the count data.
- PCA for RNA-Seq, Create Sample Level Heat Map for RNA-Seq, and Create Feature Level Heat Map for RNA-Seq automatically perform TMM normalization, followed by log2 CPM using a prior, and Z-Score normalization.
- Create Expression Browser automatically performs TMM normalization, followed by CPM without using a prior.
- Differential Expression for RNA-Seq and Differential Expression in Two Groups can perform either TMM or housekeeping gene normalization. They use the normalization factor in their statistical model: for sample
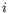 , the library size normalization factor is the
, the library size normalization factor is the
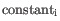 described in The GLM model.
described in The GLM model.
TMM normalization
A TMM (Trimmed Mean of M) normalization factor is computed by comparing the samples against a reference sample ([Robinson and Oshlack, 2010]). The reference is the sample whose upper-quartile (75th percentile) of counts is closest to the mean upper-quartile across all samples.
TMM normalization assumes that most genes are not differentially expressed. Therefore, it is important not to make subsets of the count data before doing statistical analysis or visualization, as this can lead to differences being normalized away.
Housekeeping gene normalization
The normalization factor for a sample is the natural logarithm of the geometric mean of the expressions of the housekeeping genes for that sample. Housekeeping genes can either be specified directly, or the most suitable subset of a short list of genes can be selected using the GeNorm algorithm [Vandesompele et al., 2002].
We recommend housekeeping genes rather than TMM normalization when:
- Working with Targeted RNA Panels.
- The TMM assumption that most genes are not differentially expressed does not hold.
Housekeeping gene normalized expressions are not directly available, but they are easy to calculate:
- Run Create Expression Browser using all samples.
- Export the total counts from the expression browser using Export table as currently shown to an .xlsx or .csv file.
- For each sample, calculate the geometric mean of the total counts of the housekeeping genes.
- Calculate the normalized expression for each gene as
total count / geometric mean. If needed, multiply by a constant factor to bring the normalized expressions to a more convenient scale.
Consider the following total counts, where HKG1 and HKG2 are housekeeping genes:
| Gene | Sample1 | Sample2 | Sample3 |
| HKG1 | 1 | 3 | 2 |
| HKG2 | 3 | 3 | 2 |
| Gene1 | 8 | 6 | 2 |
| Gene2 | 4 | 4 | 4 |
| Gene3 | 1 | 0 | 1 |
The geometric mean of the total counts of the housekeeping genes is:
| Sample1 | Sample2 | Sample3 |
|
|
The normalized expressions are then:
| Gene | Sample1 | Sample2 | Sample3 |
| Gene1 | 4619 | 2000 | 1000 |
| Gene2 | 2309 | 1333 | 2000 |
| Gene3 | 577 | 0 | 500 |
CPM
CPM (Counts Per Million) are calculated as follows, using an approach similar to EdgeR [Robinson et al., 2010]):
- If using a prior, calculate it as the scaled library size:
prior = library_size/average_library_size. Otherwise, the prior is set to 0. - Add the prior to each total count:
adjusted_count = prior + total_count. - Adjust each library size using the prior:
adjusted_library_size = 2.0 x prior + library_size. - Calculate the CPM as
adjusted_count * 1E6 / adjusted_library_size.
Z-Score normalization
Z-Score normalization performs a cross-sample normalization. For each feature (gene or transcript), the data is shifted and scaled so that the mean is zero, and the standard deviation one.