Specifying reads and reference
To start the RNA-Seq analysis, go to:
Toolbox | Transcriptomics Analysis (![]() ) | RNA-Seq Analysis (
) | RNA-Seq Analysis (![]() )
)
This opens a dialog where you select the sequencing reads. Note that you need to import the sequencing data into the Workbench before it can be used for analysis. Importing read data is described in Import Sequencing Data.
If you have several samples that you wish to analyze independently and compare afterwards, you can run the analysis in batch mode.
Click Next when the sequencing data are listed in the right-hand side of the dialog.
You are now presented with the dialog shown in figure 26.4.
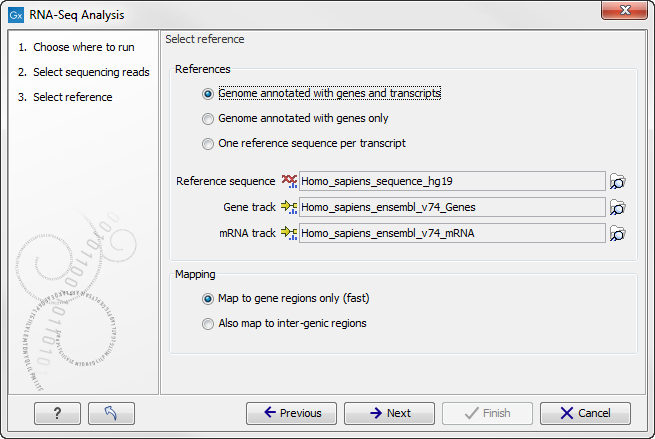
Figure 26.4: Defining a reference genome for RNA-Seq.
At the top, there are three options concerning how the reference sequences are annotated.
- Genome annotated with genes and transcripts. This option should be used when both gene and mRNA annotations are available. The mRNA annotations are used to define how the transcripts are spliced (as shown in figure 26.1). This option should be used for Eukarotes since it is the only option where splicing is taken into account. Note that genes and transcripts are linked by name only (not by position, ID etc). When this option is selected, both a Gene and an mRNA track should be provided in the boxes below. Annotated reference genomes be can obtained in various ways:
- Directly downloaded as tracks using the Download Reference Genome Data tool (see Download reference genome).
- Imported as tracks from fasta and gff/gtf files (see Import tracks)
- Imported from Genbank or EMBL files and converted to tracks (see Converting data to tracks and back).
- Downloaded from Genbank (see GenBank search) and converted to tracks (see Converting data to tracks and back).
- Genome annotated with genes only. This option should be used for Prokaryotes where transcripts are not spliced. When this option is selected, a Gene track should be provided in the box below. The data can be obtained in the same ways as described above.
- One reference sequence per transcript. This option is suitable for situations where the reference is a list of sequences. Each sequence in the list will be treated as a "transcript" and expression values are calculated for each sequence. This option is most often used if the reference is a product of a de novo assembly of RNA-Seq data. When this option is selected, only the reference sequence should be provided, either as a sequence track or a sequence list.
At the bottom of the dialog you can choose the reference content to map to. Note that this is only relevant when using an annotated reference:
- Map to gene regions only. This option will ignore all inter-genic regions in the reference. Since only genes are considered, this options is also significantly faster than the alternative option. The effect of restricting the mapping to genes only is that any reads coming from genes or transcripts that are not part of the annotations will either be unmapped or map to another transcript with a similar sequence (e.g. a pseudo-gene). For poorly annotated references, it is possible to improve the annotations using the Transcript Discovery plugin which is freely available for download in the Plugin Manager (see Installing plugins).
- Also map to inter-genic regions. This option will include the inter-genic regions as well. Please note that reads that map outside genes are counted as intergenic hits only and thus do not contribute to the expression values26.1. If a read maps equally well to a gene and to an inter-genic region, the read will be placed in the gene.
Note: If the options "One reference sequence per transcript" or "Also map to inter-genic regions" are chosen, the internal data structure used for mapping the reads can be reused, resulting in a dramatically reduced runtime. For more information about this, please see Reference caching.
Footnotes
- ... values26.1
- The reads will indirectly impact the RPKM expression values as they will be counted in the total number of mapped reads which is used to calculate RPKM (Definition of RPKM)