Significance of variant
At a given position, when the reads have been filtered, the remaining reads will be compared to the reference sequence to see if they are different at this position (for de novo assembly the consensus sequence is used for comparison). For a variant to be reported, it has to comply with the significance threshold specified in the dialog shown in figure 29.5.
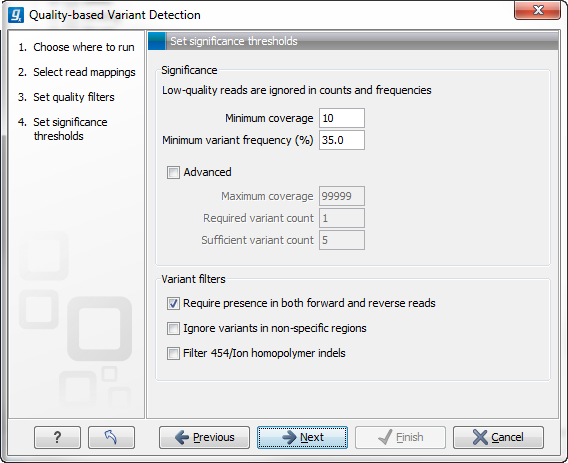
Figure 29.5: Significance thresholds.
- Minimum coverage. If variants were called in areas of low coverage, you would get a higher amount of false positives. Therefore you can set the minimum coverage threshold. Note that the coverage is counted as the number of valid reads at the current position (i.e. the reads remaining when the quality assessment has filtered out the bad ones).
- Minimum variant frequency. This option is the threshold for the number of reads that display a variant at a given position, or in other words, the reported zygosity depends on the setting of the variant frequency parameter. Setting the percentage at 35% means that at least 35% of the validated reads at this position should have a different base than the reference in order to be considered heterozygous rather than homozygous. This means that if, in one reference position, A is represented in more than 35% of the reads and C is also represented in more than 35% of the reads, the variant would be considered heterozygous because two different alleles were called for the same variant. If one of these bases (A and C in this example) is the reference base, then it will be reported in the variant track as the reference allele variant, but not in the annotated table.
- Maximum coverage. Although it sounds counter-intuitive at first, there is also a good reason to be suspicious about high-coverage regions. Read coverage often displays peaks in repetitive regions where the alignment is not very trustworthy. Setting the maximum coverage threshold higher than the expected average coverage (allowing for some variation in coverage) can be helpful in ruling out false positives from such regions. You can see the distribution of coverage by creating a Detailed mapping report.
The result table, created by the variant detection, includes information about coverage, so you can specify a high threshold in this dialog, check the coverage in the result afterwards, and then run the variant detection again with an adjusted threshold.
- Required variant count. This option is the threshold for the number of reads that display a variant at a given position. In addition to the percentage setting in the simple panel above, this setting is based on absolute counts. If the count required is set to 3, it means that even though the required percentage of the reads has a variant base, it will still not be reported if there are less than 3 reads supporting the variant.
- Sufficient variant count. This option can be used for deep sequencing data where you have very high coverage and many different alleles. In this case, the percentage threshold is not suitable for finding valid variants only present in a small number of alleles. If the sufficient variant count is set to 5, it means that as long as there are 5 reads supporting a variant, it will be called irrespective of the frequency setting (it still has to be above the required variant count which should always be lower than the sufficient variant count).
When there are ambiguity bases in the reads, they will be treated as separate variants. This means that e.g. a Y will not be collapsed with C or T in other reads. Rather, the Ys will be counted separately.
Variant filters
Below the significance settings, there are filters that can be useful for removing false positives:
- Require presence in both forward and reverse reads. Some systematic sequencing errors can be triggered by a certain combination of bases. This means that sequencing one strand may lead to sequencing errors that are not seen when sequencing the other strand (see [Nguyen et al., 2011] for a recent study with Illumina data). This can easily lead to false positive variant calls, and by checking this filter, the minimum ratio between forward and reverse reads supporting the variant should be at least 0.05. In this way, systematic sequencing errors of this kind can be eliminated. The forward/reverse reads balance is also reported for each variant in the result (see Variant data).
- Ignore variants in non-specific regions. Variants in regions covered by one or more non-specific reads are ignored.
- Filter 454/Ion homopolymer indels. The 454 and Ion Torrent/Proton sequencing platforms exhibit weaknesses when determining the correct number of the same kind of nucleotides in a homopolymer region (e.g. AAA). This leads to a high false positive rate for calling indels in these regions. This filter is very basic: it removes all indels that are found within or just next to a homopolymer region. A homopolymer region is defined as at least two consecutive identical bases in the reference.