Calculating expression values from RNA-Seq
When the reference has been defined, click Next and you are presented with the dialog shown in figure 28.8.
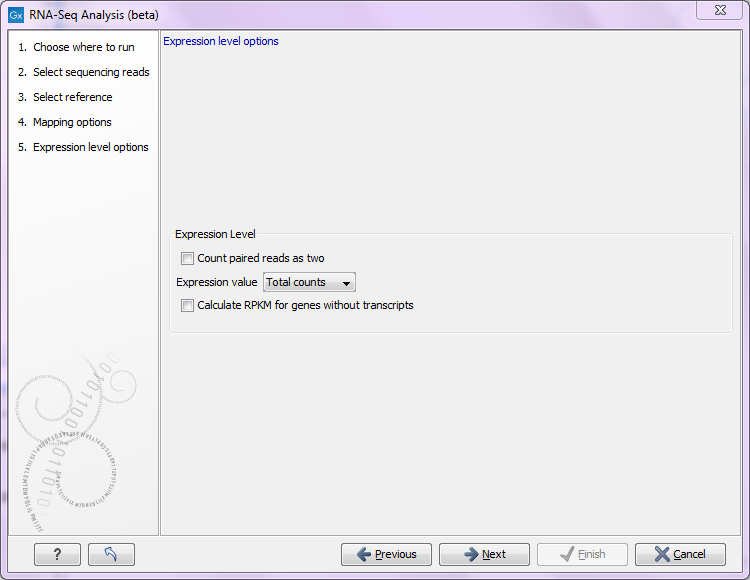
Figure 28.8: Defining how expression values should be calculated.
These parameters determine the way expression values are counted. Some background information on how paired reads are handled is useful before describing the parameters.
Paired reads in RNA-Seq
The CLC Genomics Workbench supports the direct use of paired data for RNA-Seq. A combination of single reads and paired reads can also be used. There are three major advantages of using paired data:
- Since the mapped reads span a larger portion of the reference, there will be fewer non-specifically mapped reads. This means that generally there is a greater accuracy in the expression values.
- This in turn means that there is a greater chance of accurately measuring the expression of transcript splice variants. As single reads (especially from the short reads platforms) typically only span one or two exons, many cases will occur where expression of splice variants sharing the same exons cannot be determined accurately. With paired reads, more combinations of exons will be identified as being unique for a particular splice variant.28.2
- It is possible to detect Gene fusions when one read in a pair maps in one gene and the other part maps in another gene. Several reads exhibiting the same pattern is supporting the presence of a fusion gene.
You can read more about how paired data are imported and handled in General notes on handling paired data.
When counting the mapped reads to generate expression values, the CLC Genomics Workbench needs to be told how to handle the counting of paired reads. The default behavior of the CLC Genomics Workbench is to count fragments rather than individual reads when two reads map as an intact pair. That is, an intact pair is given a count of one. Reads from a pair are considered part of a broken pair when the reads map outside the estimated pair distance, either map in wrong orientation or only one of the reads of the pair map. Neither member of a broken pair are counted when the default counting scheme is used. The reasoning is that when reads map as a broken pair, it is in indication that something is not right. For example, perhaps the transcripts are not represented correctly on the reference or there are errors in the data. In general, more confidence can be placed on an intact pair representing transcription within the sample. If a combination of paired and single reads are input into the analysis, then single reads that map are given a count of one. This is different from reads input into the analysis as part of a pair, but where their partner did not map.
In some situations it may be too strict to disregard broken pairs as is done using the default counting scheme. This could be the case where there is a high degree of variation in the sample compared to the reference or where the reference lacks comprehensive transcript annotations. By checking the Count paired reads as two option, you choose to count mapped 'reads' rather than mapped 'fragments. That means that, the two reads in an intact pair are each counted as one mapped read (so an intact pair contributes with a total count of two), and mapped members of broken pairs will each get given a count of one. Single mapped reads are also given a count of one. Note that this approach does not represent the abundance of fragments being sequenced correctly, since the two reads of a pair derive from the same fragment, whereas a fragment sequenced with single reads only give rise to one read.
When looking at the mappings, reads from broken pairs have a darker color than reads that are intact pairs or originally single reads.
Expression value
The expression values are created on two levels as two separate result files: one for genes and one for transcripts (if the "Genome annotated with genes and transcripts" is selected in figure 28.4). The content of the result files is described in RNA-Seq results.
The Expression value parameter describes how expression per gene or transcript can be defined in different ways on both levels:
- Total counts. When the reference is annotated with genes only, this value is the total number of reads mapped to the gene. For un-annotated references, this value is the total number of reads mapped to the reference sequence. For references annotated with transcripts and genes, the value reported for each gene is the number of reads that map to the exons of that gene. The value reported per transcript is the total number of reads mapped to the transcript.
- Unique counts. This is similar to the above, except only reads that are uniquely mapped are counted (read more about the distribution of non-specific matches in Defining mapping options for RNA-Seq).
- RPKM. This is a normalized form of the "Total counts" option (see more in Definition of RPKM).
Please note that all values are present in the output. The Expression value in this dialog is solely used to inform the Workbench about which expression value should be applied when using the result in downstream analysis.
For genes without annotated transcripts, the RPKM cannot be calculated since the total length of all exons is needed. By checking the Calculate RPKM for genes without transcripts, the length of the gene will be used in place of an "exon length". If the option is not checked, there will be no RPKM value reported for those genes.
Genes in Operons
In the case of operons, where several genes are transcribed in the same mRNA transcript and are therefore located directly alongside each other, it is likely that some RNA-seq reads will map across the boundary of two different genes. If the mapping option "Also map to inter-genic regions" has been turned on, then reads like this will not be counted towards expression of any gene. If the option "Map to gene regions only (fast)" has been chosen, then reads will be counted towards only one gene within the operon: the one it mapped to best, which will usually mean the gene which the longest segment of the read mapped to. This means that the value for the expression of a particular operon in the RNA-seq results will be divided across the component genes of that operon.
Footnotes
- ... variant.28.2
- Note that the CLC Genomics Workbench only calculates the expression of the transcripts already annotated on the reference.