Whole Transcriptome Sequencing (WTS)
The technologies originally developed for next-generation DNA sequencing can also be applied to deep sequencing of the transcriptome. This is done through cDNA sequencing and is called RNA sequencing or simply RNA-seq.
One of the key advantages of RNA-seq is that the method is independent of prior knowledge of the corresponding genomic sequences and therefore can be used to identify transcripts from unannotated genes, novel splicing isoforms, and gene-fusion transcripts [Wang et al., 2009,Martin and Wang, 2011]. Another strength is that it opens up for studies of transcriptomic complexities such as deciphering allele-specific transcription by the use of SNPs present in the transcribed regions [Heap et al., 2010].
RNA-seq-based transcriptomic studies have the potential to increase the overall understanding of the transcriptome. However, the key to get access to the hidden information and be able to make a meaningful interpretation of the sequencing data highly relies on the downstream bioinformatic analysis.
In this chapter we will first discuss the initial steps in the data analysis that lie upstream of the analysis using ready-to-use workflows. Next, we will look at what the individual ready-to-use workflows can be used for and go through step by step how to run the workflows.
CLC Workbench offers a range of different tools for RNA-seq analysis. Currently 5 different ready-to-use workflows for 3 different species - human (![]() ), mouse (
), mouse (![]() ) and rat (
) and rat (![]() ) - are available for analysis of RNA-seq data:
) - are available for analysis of RNA-seq data:
- Annotate Variants (WTS)
- Compare Variants in DNA and RNA
- Identify Candidate Variants and Genes from Tumor Normal Pair
- Identify Variants and Add Expression Values
- Identify and Annotate Differentially Expressed Genes and Pathways
The ready-to-use workflows can be found in the toolbox under Whole Transcriptome Sequencing as shown in figure 16.1.
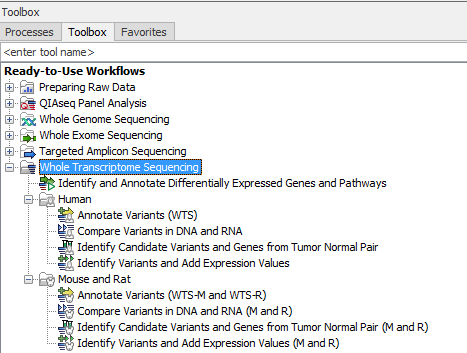
Figure 16.1: The RNA-seq ready-to-use workflows.
Remember you will have to prepare data with the Prepare Raw Data workflow described in Preparing Raw Data before you proceed to.
Also note that in case of workflows annotating variants using databases available for more than one population, you can select the population that matches best the population your samples are derived from.
To get the most out of the RNA-Seq analysis tools of the workbench, we recommend that all input expression tracks have associated metadata. For information about how to use and setup metadata, please see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Metadata.html.
Subsections
- Annotate Variants (WTS)
- Compare Variants in DNA and RNA
- Identify Candidate Variants and Genes from Tumor Normal Pair
- Identify Variants and Add Expression Values
- Identify and Annotate Differentially Expressed Genes and Pathways