Trim Primers and their Dimers of Mapped Reads
The Trim Primers and their Dimers of Mapped Reads tool is used in the QIAGEN GeneRead Panel Analysis ready-to-use workflow. It is also provided as a separate tool to be used for targeted amplicon sequencing experiments with many targets (and as a consequence many primers). To be able to trim off the primers used in your sequencing experiment you must know the primer sequences as you will need to specify which target primer sequence file to use. To learn how to import descriptions of primer locations from a generic text format file or from a QIAGEN gene panel primer file, see the description of the Import Primer Pairs tool here: http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Import_Primer_Pairs.html.
The Trim Primers and their Dimers of Mapped Reads not only trims off primers but also makes use of the primer pairs in the trimming process to predict and trim possible primer dimerizations. The prediction is based on the primer pairs, the reference, and user settings that are described later in this section. Removal of primers and their dimers from the mapped reads ensures that no bias is introduced in the variant calling as would be the case if the primers and dimers were considered to be part of the sequencing reads.
When running the Trim Primers and their Dimers of Mapped Reads tool, primers of the reads are trimmed first. The tool then looks for primer dimerization artifacts and trim them.
Primer trimming
The user can specify the fraction of the primer that must overlap with a read's aligned bases in order to record a primer hit.The Trim Primers and their Dimers of Mapped Reads is strict regarding primer position, i.e., if a primer begins after the read at the 5' end, it will not be considered for trimming. The read's unaligned bases are taken into account. For example, if the primer begins two positions before the read's first aligned base and the read has three unaligned bases, the primer is said to begin after the read. Similarly, a primer that ends before the read at the 3' end is not considered. Again unaligned bases are taken into account.
Primer dimer trimming
The primer dimer trimming is done in two steps.In the first step, all primers are compared against each other for possible primer dimerization. The user may specify the minimum number of bases that needs to bind for primers to dimerize and amplify. After the first step, a list of possible primer dimerizations have been compiled for each primer.
In the second step, the actual trimming is performed. All reads are examined, and if the read was trimmed by a primer, p, and the read starts with the sequence predicted by one of p's possible primer dimerizations, it is assumed that the read has a primer-dimer artifact. The tool proceeds to trim the read so the artifact is unaligned. In the case where the read only consists of the primer-dimer artifact sequence, the read will be discarded.
Running the tool
The Trim Primers and their Dimers of Mapped Reads can be found in the toolbox:
Toolbox | Resequencing Analysis (![]() ) | Trim Primers and their Dimers of Mapped Reads (
) | Trim Primers and their Dimers of Mapped Reads (![]() )
)
In the first wizard step (figure 10.12), you are asked to select the read mapping. If you would like to analyze more than one read mapping, you can choose to run the analysis in batch mode by ticking the "Batch" box in the lower left corner of the wizard and then selecting the folder that hold the read mappings you want to analyze.
Figure 10.12: Select files to import.
In the next wizard step (see figure 10.13), specify the parameters for the trimming tool.
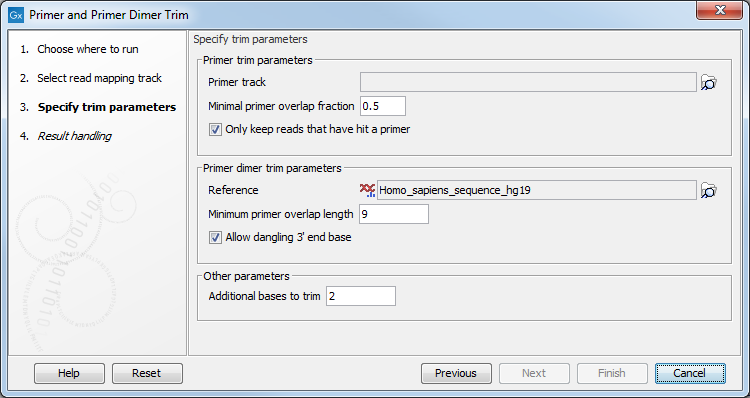
Figure 10.13: Select your primer location file and choose whether you want to keep or discard reads with no matching primers.
- Primer trim parameters
- Primer track Click on the folder icon on the right-hand side of the wizard to select your primer location file.
- Minimal primer overlap fraction Specifies the fraction of the primer that must overlap with the read's aligned bases in order to record a primer hit. Setting the fraction to 0.0 will disable this requirement.
- Read handling configuration If you tick "Only keep reads that have hit a primer", reads with no matching primers will be discarded.
- Primer dimer trim parameters
- Reference Click on the folder icon on the right-hand side of the wizard to select your reference location file.
- Minimum primer overlap length The minimum number of bases that needs to bind for primers to dimerize and amplify.
- Allow dangling 3' end base If you tick "Allow dangling 3' end base", a mismatch is allowed in the primer dimerization at the 3' end.
- Other parameters
- Additional bases to trim This number of nucleotides will be trimmed off a read right after the primer. This trimming is not done on reads for which primer dimer artifacts were identified. This is set by default to 2 to avoid false positive calls and increase accuracy of the coverage calculation in the report.
In the last wizard window, choose to save the result of the primer trimming and click Finish.
Output of the Trim Primers and their Dimers of Mapped Reads tool
The output is a read file from which primers and their dimers have been trimmed off. The name of the output is the one from the original input file with "trimmed reads" appended to it. In the last wizard step it is also possible to save a track with the primer dimers that were used to trim reads. The track contains information on why the primer dimer was predicted and the number of times it was used to partially trim a read or remove a read. A read is removed if the read only consists of the primer dimer.