Identify MLST
This section describes how to perform the identification of the MLST from a genome or a read mapping and given a MLST scheme. The Identify MLST tool requires an exact match between the allele sequences in the MLST scheme and a consensus sequence. If a read mapping is used as input, a consensus of the read mapping will be used. This stringency level is needed because the alleles used for multi locus sequence typing are very similar, often almost identical. Therefore MLST typing relies on a MLST scheme that represent the organism under investigation well.
To MLST type a consensus sequence or a read mapping, go to:
Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | NGS-MLST (
) | NGS-MLST (![]() ) | Identify MLST (
) | Identify MLST (![]() )
)
In the first wizard window you are asked to select the consensus sequence or read mapping to be used for MLST typing (figure 13.3).
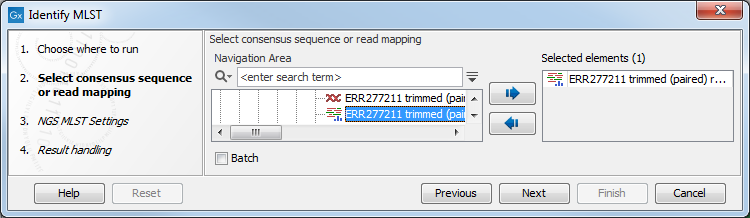
Figure 13.3: Select consensus sequence or read mapping to be used for MLST typing.
In the next window, two options should be specified (figure 13.4):
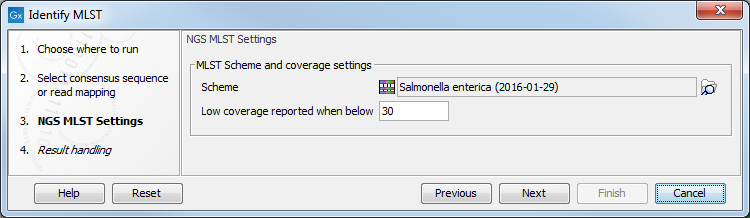
Figure 13.4: Select relevant MLST scheme(s) for typing.
- Scheme The NGS-MLST typing tool requires defined fragments of multiple housekeeping genes in the format of a MLST scheme, which should specified as a parameter (figure 13.4).
- Low coverage reported when below enable the user to specify the minimum coverage threshold. If the coverage within a found loci falls below this threshold, a warning will be printed in the output report
The tool generates two output files: the Consensus NGS MLST Report and the NGS MLST Annotation Track. The Consensus NGS MLST Report includes information on which MLST scheme was applied, the identified sequence type (ST) as well as an overview table which summarizes the targeted genes, their consensus sequence profile and coverage. The NGS MLST Annotation Track specifies the individual target regions in the coordinate system of the original input sequence.
In situations where not all (potentially no) loci are found in the consensus sequence, and a loci positions is transferable from the read mapping reference sequence, it will be reported as a possible new loci at the transferred position.
To add the obtained ST to your Result Metadata Table, see the section Add to Result Metadata Table. Note that results are added automatically to the Result Metadata Table when using the templates Type a Known Species and Type among Multiple Species workflows or their customized versions.