Create Large MLST Scheme (beta)
The Create Large MLST Scheme tool can be used to create a scheme from scratch.
To run the Create Large MLST Scheme (beta) tool choose:
Microbial Genomics Module (![]() ) | Databases (
) | Databases (![]() ) | Large MLST (
) | Large MLST (![]() ) | Create Large MLST Scheme (beta) (
) | Create Large MLST Scheme (beta) (![]() )
)
As input, the tool requires a set of complete isolate genomes in the form of one or more sequence lists or sequences. These genomes must be annotated with coding region (CDS) annotations. If these are not available, the Find Prokaryotic Genes tool (see Find Prokaryotic Genes) can be used to predict and annotate the coding regions.
Note that when working with genomes that span several chromosomes or several contigs, it is necessary to declare which sequences belong to the same genome. This is done by ensuring all sequences from the same genome have the same 'Assembly ID' and 'Latin name' annotations. Some tools, such as the Download Microbial Reference Database, will automatically assign these annotations, for a manual assignment of Assembly ID annotations, plase see Using the Assembly ID annotation.
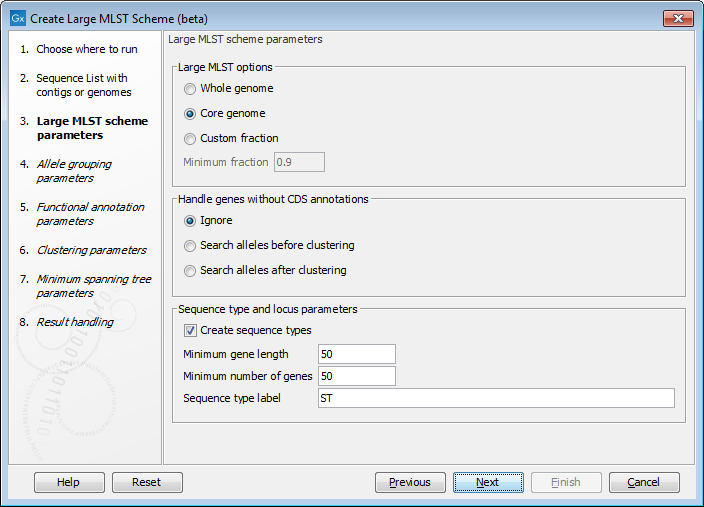
Figure 18.1: Basic options for creating a large mlst scheme.
After specifying the input, the second step is to set up the basic Large MLST Scheme creation parameters (figure 18.1).
The Create Large MLST Scheme tool works by extracting all annotated coding sequences (CDS) and clustering them into similar gene classes (loci). It is possible to specify whether we are interested in the genes that are present in some genomes (Whole genome - must be present in at least 10% of all genomes), most genomes (Core genome - must be present in at least 90% of the genomes), or a user-specified Minimum fraction.
The best results are obtained by supplying genomes with proper CDS annotations. The Handle genes without annotations option controls how genomes without CDS annotations are handled:
- Ignore: No checks are performed.
- Search alleles before clustering: All of the input genomes are blasted (using DIAMOND) against the set of annotated genes, and any new genes will be added as alleles. This is a very slow, but thorough check.
- Search alleles after clustering: After clustering the genes, all of the input genomes are blasted (using DIAMOND), but only against the longest protein in each cluster.
The Sequence type and locus parameters control the following filtering and naming options:
- Minimum gene length: the minimum length of the gene (CDS annotation) before including it in the scheme creation.
- Minimum number of genes: the minimum number of genes (CDS annotations) necessary for a given input genome, before it is included in the scheme creation.
- Sequence type label: a text label that is used as the base name of the sequence types in the scheme.
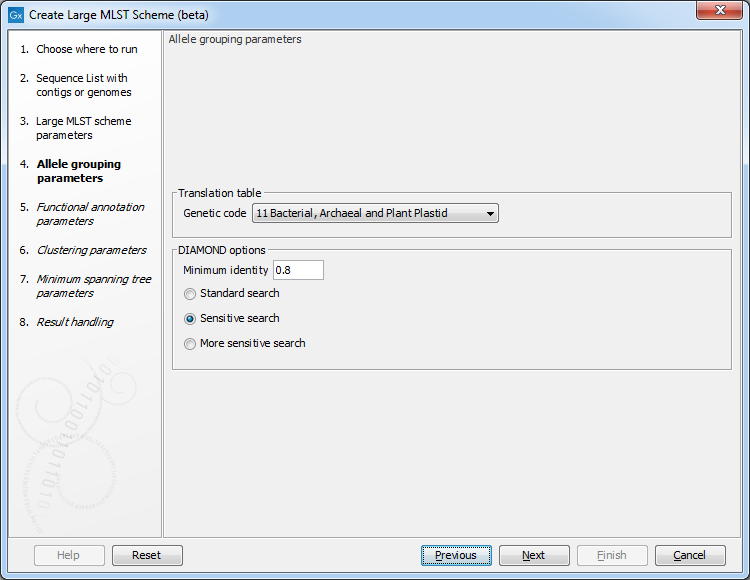
Figure 18.2: The allele grouping (clustering) options.
The Allele grouping parameters step specifies how the different genes (CDS annotations) are compared to each other. Diamond is used for this clustering. It is also possible to specify the Genetic code for the input samples.
The Minimum identity determines the minimum sequence identity before grouping protein sequences. It is also possible to specify the sensitivity of the search (Standard search, Sensitive search, and More sensitive search) - increasing the sensitivity makes the search more thorough, but also much slower. The default for this parameter is Sensitive search.
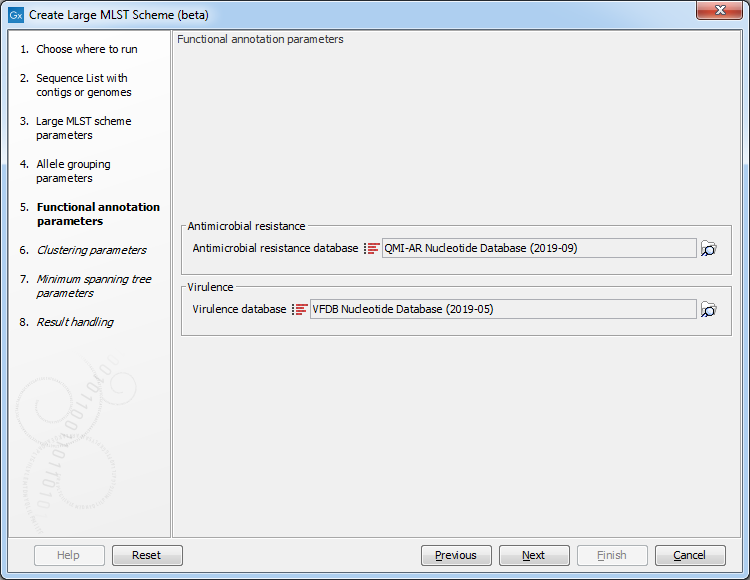
Figure 18.3: The functional annotation parameters.
It is possible to decorate the alleles with information about virulence or resistance. The information can be extracted from either a ShortBRED Marker database or a Nucleotide database. These databases can be accessed using the Download Resistance Database tool (e.g. QMI-AR for resistance or VFDB for virulence) and can be provided as input to the Create Large MLST Scheme tool at this step.
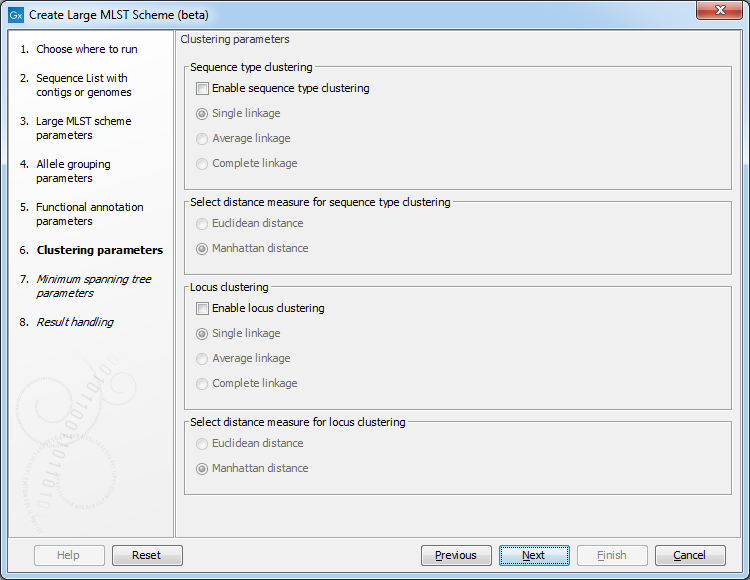
Figure 18.4: The clustering parameters.
The clustering parameters determine how the heatmap should be clustered. The heatmap cell values are the observed frequencies of a given allele compared to the other alleles in the same locus.
The possible cluster linkages are:
- Single linkage. The distance between two clusters is computed as the distance between the two closest elements in the two clusters.
- Average linkage. The distance between two clusters is computed as the average distance between objects from the first cluster and objects from the second cluster.
- Complete linkage. The distance between two clusters is computed as the distance between the two farthest objects in the two clusters.
The possible distance measures are:
- Euclidean distance: the square-root of the sum-of-square differences between coordinates.
- Manhattan distance: the sum of absolute differences between coordinates.
Note that for schemes with thousands of sequence types, the clustering may become very slow and time-consuming.
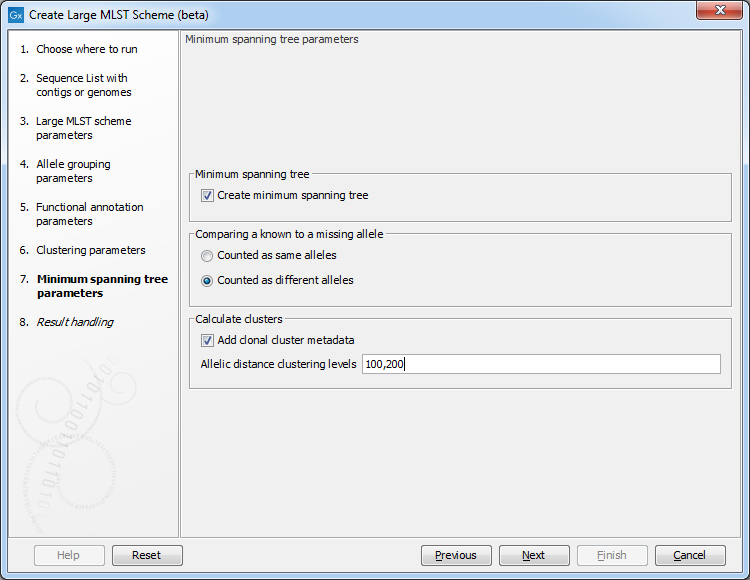
Figure 18.5: The minimum spanning tree parameters.
The following options are available when creating a minimum spanning tree:
- Comparing a known to a missing allele: the minimum spanning tree is created using a distance matrix, where the distance is calculated between all pairs of sequence types. The distance is calculated as the number of loci where the allele assignment differs. But in some cases, a locus for a sequence type may not have an assigned allele (for instance, for the accessory genes in a wgMLST scheme). If this is the case, the behavior depends on this setting: if 'counted as same alleles' is selected, a locus where at least one allele is missing for the pair being compared will be ignored (it will not count as a difference). On the other hand, if 'Counted as different alleles' is selected, a missing allele being compared to a known allele will increase the distance between the sequence types being compared.
- Add clonal cluster metadata: it is possible to assign cluster information to the scheme which will show up as metadata. The clustering is based on the minimum spanning tree, and will be similar to the clustering obtained by using the 'collapse branches' slider in the minimum spanning tree view - that is, the clustering will be single-linkage clustering - i.e. all nodes in cluster are within the specified threshold to at least one other node in the cluster. Each cluster will get a name chosen from the sequence type in the cluster with most connections.
- Allelic distance clustering levels: specifies the level at which the clustering will be performed. It is possible to specify multiple, comma-separated values. E.g. '100,200' will assign clusters at allelic distances of 100 and 200 - this will create two new metadata columns, cc_100 and cc_200 with the new cluster information.