QIAseq Pro Fastq to Germline CNV Control
The QIAseq Pro Fastq to Germline CNV Control template workflow produces coverage tables that can be used as controls for copy number variant detection.
Use the workflow to generate coverage tables for the QIAseq Pro Fastq to Annotated Germline Variants (QIAseq Pro Fastq to Annotated Germline Variants) template workflow.
QIAseq Pro Fastq to Germline CNV Control can be found at:
Template Workflows | LightSpeed Workflows (![]() ) | QIAseq workflows (
) | QIAseq workflows (![]() ) | QIAseq Targeted DNA Pro (
) | QIAseq Targeted DNA Pro (![]() ) | QIAseq Pro Fastq to Germline CNV Control (
) | QIAseq Pro Fastq to Germline CNV Control (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the first wizard step, select a Reference Data Set (figure 6.13).
If you have not downloaded the Reference Data Set yet, the dialog will suggest the relevant data set and offer the opportunity to download it using the Download to Workbench button.
This workflow has been set up to process data generated with QIAseq Targeted DNA Pro panels, and it is important to choose the right reference data to get the reads correctly processed.
The off-the-shelf QIAseq Targeted DNA Pro panels are available in the QIAseq DNA Pro Panels hg38 reference data set. If you have not downloaded the Reference Data Set yet, the dialog will offer the opportunity to download it using the Download to Workbench button.
If the QIAseq DNA Pro Panels hg38 reference data set does not contain the needed primers and target regions, a custom reference data set can be created, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Custom_Sets.html.
The reference data set for QIAseq Targeted DNA panels should not be used with this workflow. The differences in read structure will for example prevent primers from being correctly trimmed.
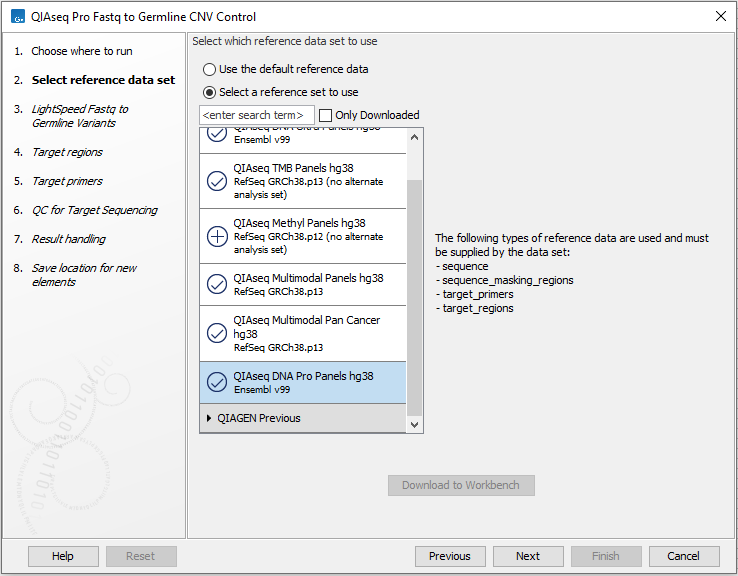
Figure 6.58: Select a reference data set.
In the LightSpeed Fastq to Germline Variants wizard step (figure 6.14) you have the following options:
- Reads (fastq) Press Browse to select fastq files for analysis.
- Masking mode To enable reference masking when mapping reads, set this option and select a masking track.
- Masking track Provide a masking track for the chosen reference genome if reference masking has been enabled.
- Batch Select if fastq files from different samples are used as input, and each sample should be analyzed individually. The names of the fastq files must follow standard Illumina naming scheme to allow the tool to identify individual fastq files as belonging to the same sample.
- Join lanes when batching Select to join fastq files from the same sample that were sequenced on different lanes.
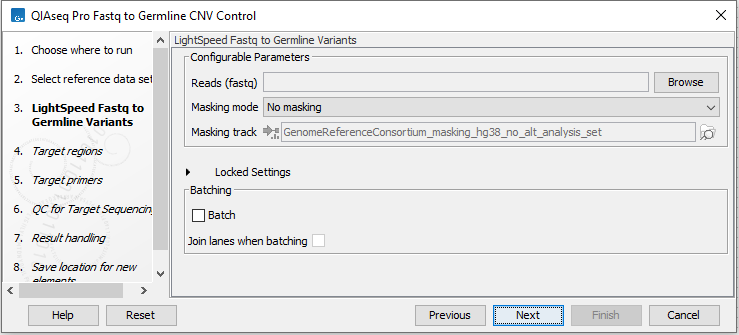
Figure 6.59: Select fastq files.
In the next dialog (figure 6.15), specify the relevant target regions from the drop down list.
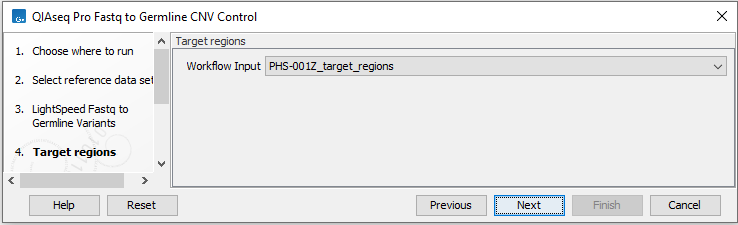
Figure 6.60: Select target regions.
Repeat the selection of the appropriate track for Target primers in the subsequent dialog (figure 6.16).
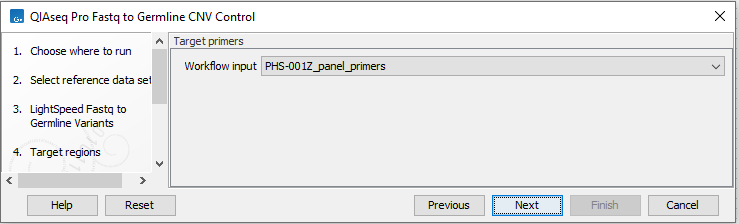
Figure 6.61: Select target primers.
In the dialog called QC for Target Sequencing, you can modify the Minimum coverage needed on all positions in a target for this target to be considered covered (figure 6.17). Note that the default value for this tool depends on the application chosen (somatic or germline).
Figure 6.62: Set the Minimum coverage parameter of the QC for Target Sequencing.
In the final wizard step, choose to Save the results of the workflow and specify a location in the Navigation Area before clicking Finish.
Subsections