Tracks
The primary outputs of the Transcript Discovery tool are a gene and a transcript tracks containing all "known" genes/transcripts (if these were supplied as input), as well as some "unknown" genes/transcripts (i.e., those that we have predicted, unless none were predicted). Note that by definition, every unknown gene/transcript present in the track is detected, while "known" genes/transcripts may or may not be detected.
Predicted gene track
A Predicted gene track viewed a table is shown in figure 3.7.
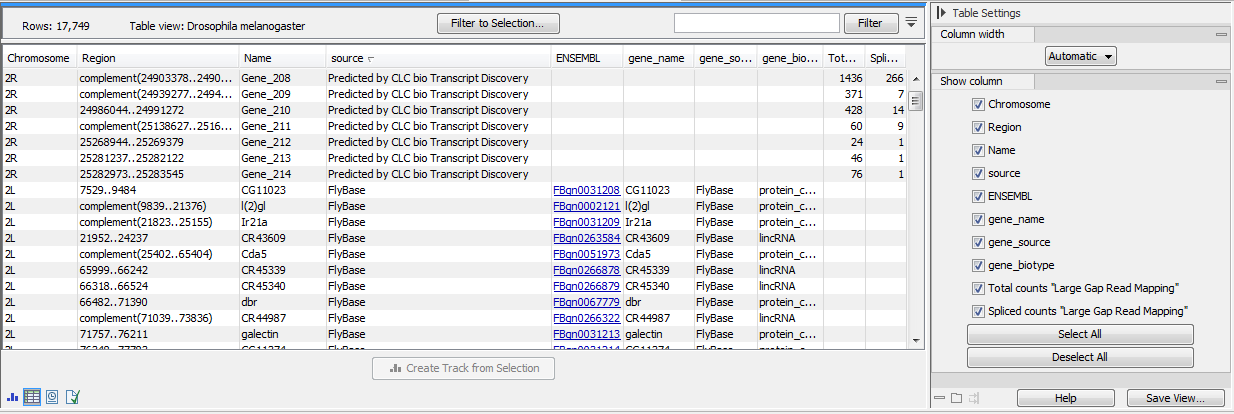
Figure 3.7: A Predicted gene track seen as a table, and showing "known" and "unknown" genes.
The column headers of the Predicted gene table are:
- Chromosome
- Region
- Name
- source. Every unknown gene or transcript will be described as "Predicted by CLC bio Transcript Discovery".
- ENSEMBL. Links to the ENSEMBL database for "known" genes.
- gene_name
- gene_source
- gene_biotype
- Total counts "sample name"
- Spliced counts "sample name"
Unknown genes are given names of the form Gene_1, Gene_2 etc. In order to avoid name clashes with previous predictions, the algorithm checks the previously annotated transcripts for genes with names of this form. New predictions will then be output with the next available index. For example, if a "Gene_11" is already present in the previously annotated transcripts, then the first new gene will be Gene_12.
These annotations sometimes include the sample name, such that if the tool is run on "Sample A", then the output transcript track will have table columns of the form "Coverage "Sample A"". This new track can then be supplied as input when the tool is run on "Sample B". The output transcript track will then have table columns "Coverage "Sample A"" and "Coverage "Sample B"".
Predicted transcript track
Similarly a transcript track contains all the "known" input transcripts plus "unknown" transcripts if any were detected (figure 3.8).
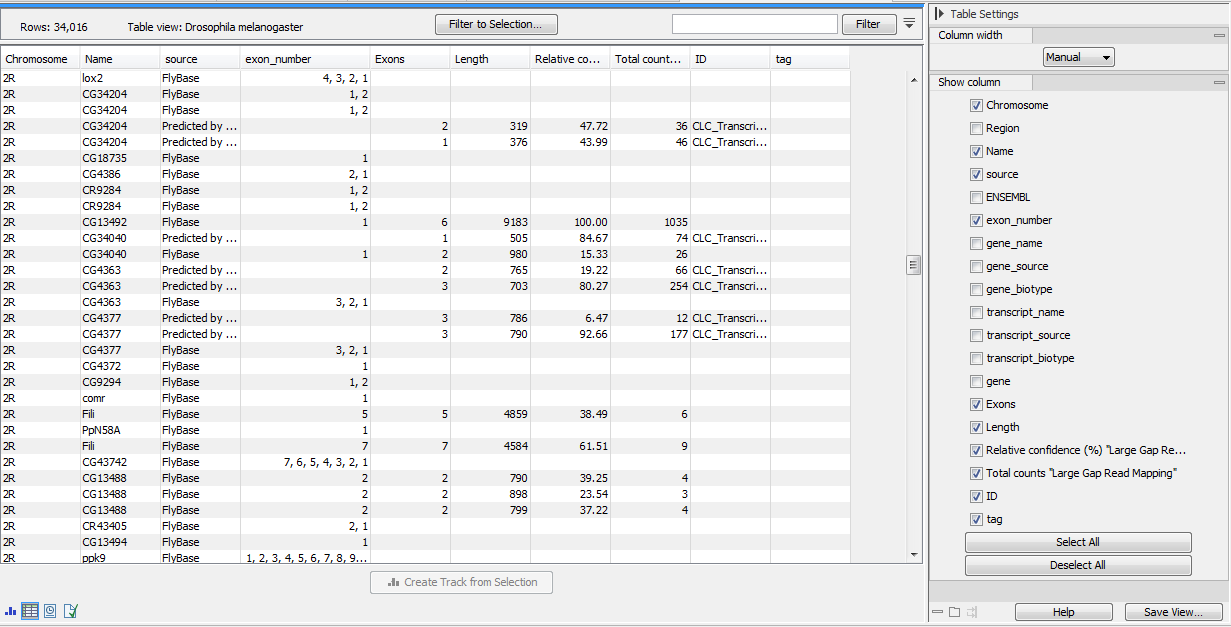
Figure 3.8: A Predicted transcript track seen as a table, and showing "known" and "unknown" transcripts.
For detected transcripts, the following annotations are available:
- Chromosome
- Region
- Name. Unknown transcripts wear the name of their gene, which guarantees that they can be linked with the gene in the RNA-Seq Analysis tool. The gene name for a particular transcript is extracted from the GFF3 parent/child relation if available (see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=GFF3_format.html to learn more about GFF3 format).
- source. Every unknown gene or transcript will be described as "Predicted by CLC bio Transcript Discovery".
- gene
- Exons
- Length
- Relative confidence (%) "sample name". The abundance of a transcript: % of transcripts for a given gene that are expected to come from that transcript.
Note that observed total relative confidence for all reported transcripts for a given gene can add up to less than 100%, as transcripts below 5 The relative confidence does not use the % of reads, as a long transcript will produce more reads than a short transcript.
- Total counts "sample name"
- ID
Predicted CDS track
Predicted CDS tables have, on top of some of the column headers described above, the following ones:- codon_start
- Parent. This is the ID of the corresponding predicted transcript.
Note that uncertainty in whether an ORF might start before the first observed start codon can be seen by the use of an "open" position, for example <130 for an ORF that is annotated at position 130, but may actually begin earlier in the reference figure 3.9
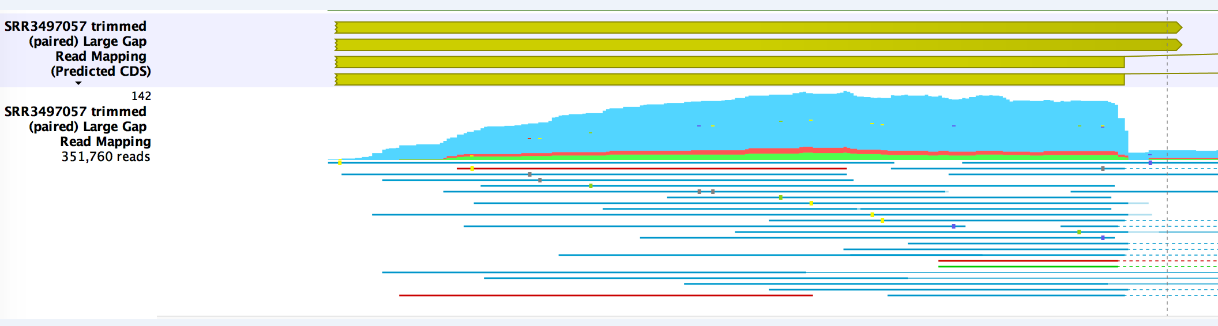
Figure 3.9: A jagged line at the beginning of the annotation shows that the ORF may start before the first observed start codon.
Accepted events track
Accepted events tables have, on top of some of the column headers described above, the following ones:- Evidence for transcripts
- Boundaries moved based on nearby events
- Spliced evidence
- Coverage "sample name"
Rejected events track
Rejected events tables have, on top of some of the column headers described above, the following ones:- Name, which describes which filter caused an event to be excluded from the final set of predicted transcripts (see figure 3.10)
- Unspliced counts "sample name"
- Max intron length
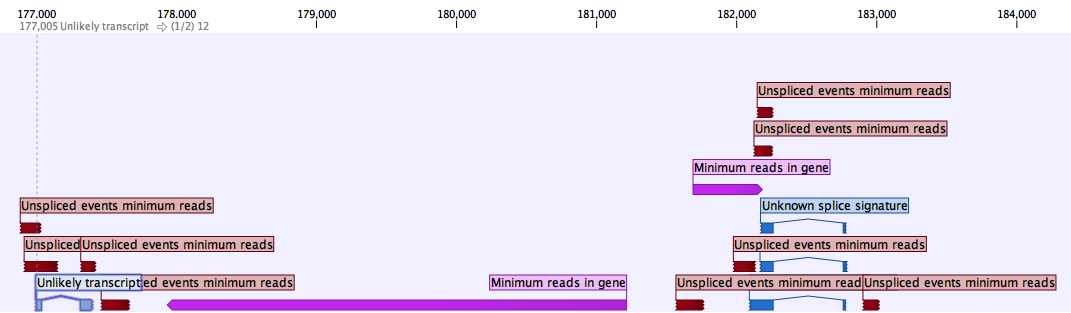
Figure 3.10: Rejected events' Names as seen in the track view of the Rejected events track.
The Name describing the filter causing an event to be rejected also refers to which filter can be adjusted in a second iteration of the tool to report the event in the Accepted events track:
- Event filters can be configured differently to accept the following rejected events:
- Minimum spliced reads
- Minimum spliced coverage ratio
- Unknown splice signature
- Chimeric reads
- Unspliced events minimum reads
- Unspliced events minimum coverage ratio
- Gene filters can be configured differently to accept the following rejected events:
- Gene without spliced reads
- Minimum reads in gene
- Minimum predicted gene length
However, there are no configurable filters to change the rejection of the following events:
- Unlikely transcript. The event is only present in transcripts that are estimated to make up <5
- Unspliced event inside. intron The event is located entirely within introns of spliced events, and does not overlap with other unspliced events. It may be transcriptional noise.
- Low coverage outside spliced region The event is at the boundaries of a gene and has <25
Track list
It can be very handy to see annotations reflecting the various steps in the transcript discovery in a Track list. An example is shown in figure 3.11
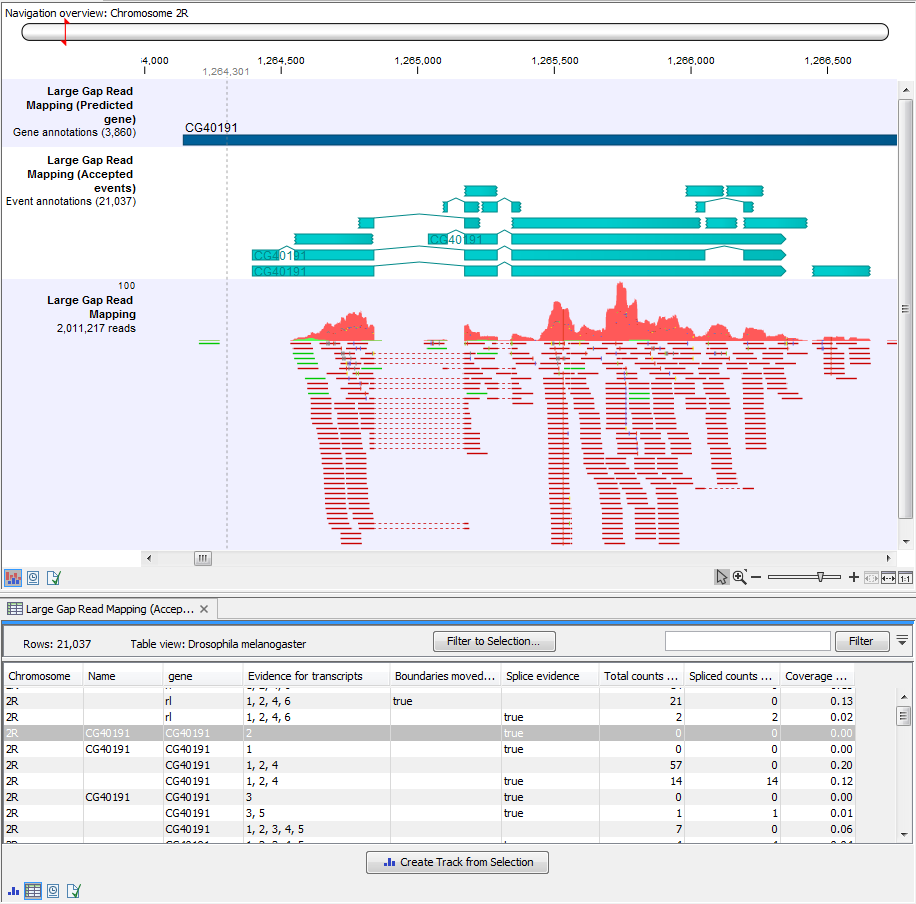
Figure 3.11: A track list showing the read mapping along with the Accepted events track and the Predicted gene track.