Large Gap Read Mapping
The Large Gap Read Mapping tool maps reads to a reference, while allowing for large gaps in the mapping. It is developed to support transcript discovery using RNA-Seq data, since it is able to map RNA-Seq reads that span introns without requiring prior transcript annotations.The Large Gap Read Mapping tool works by iteratively applying the standard read mapper of CLC Genomics Workbench to each read as follows:
- Find the best match for the read.
- If the match is good enough (according to the settings, see below), the read is mapped to this position.
- If there is an unaligned end which is long enough for the mapper to handle (15 bp for standard mapping), this part of the read is used as input to step 1.
- This continues until no reads have unaligned ends that are longer than 15 bp. The number of rounds required scales with the length of the reads. For short 100 bp reads the maximum will usually be maximum three rounds of mapping (corresponding to spanning two introns). For full-length transcripts more than ten rounds may be required.
The matched region of the read identified in the first round of the mapping is called the seed segment (or just 'seed'). Matched regions found in later rounds are called non-seed segments.
To run the Large Gap Read Mapping tool, go to:
Tools | RNA-Seq Analysis (![]() ) | Transcript Discovery (
) | Transcript Discovery (![]() ) | Large Gap Read Mapping
) | Large Gap Read Mapping
First specify the RNA-Seq reads that should be analyzed (figure 2.1):
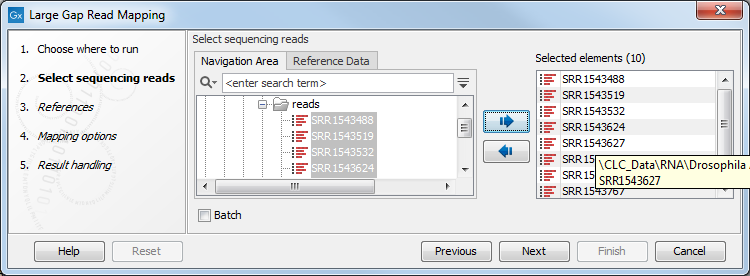
Figure 2.1: Selecting input reads for the Large Gap Read Mapping tool.
In the next dialog, specify the reference (a sequence track or a sequences list) to which the reads should be mapped (figure 2.2):
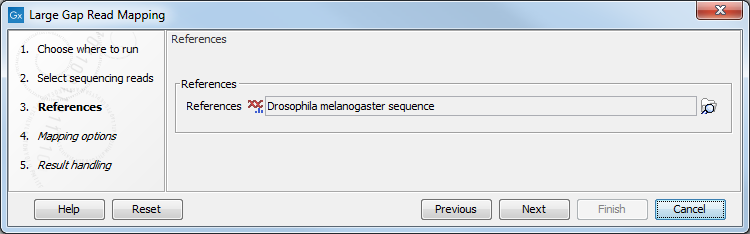
Figure 2.2: Selecting references for the Large Gap Read Mapping tool.
In the Mapping options dialog, specify the following parameters (figure 2.3):
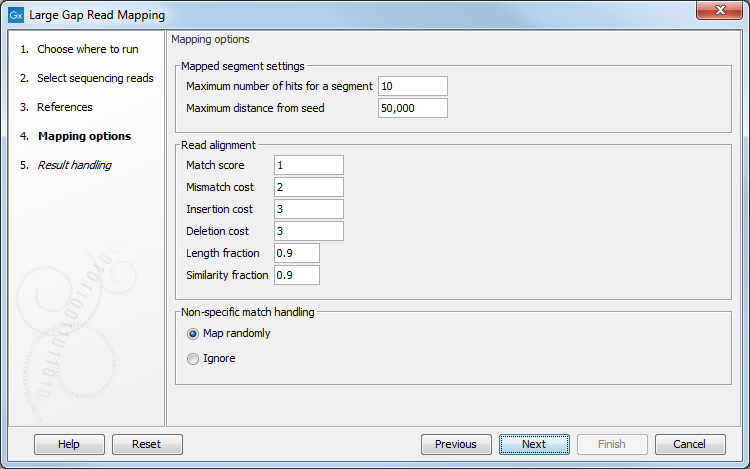
Figure 2.3: Specify the parameters for the Large Gap Read Mapping tool.
- Maximum number of hits for a segment is the maximum number of hits that a segment is allowed to have in order for the read to be mapped. If, for a non-seed segment, this number is exceeded, the read is classified as unmapped. If it is not exceeded, all the multiple hit positions will be considered. If the seed makes up the full read it may map in up to 'Maximum number of hits' positions.
- Maximum distance from seed is the maximum distance allowed between the first match part (the seed) and any subsequent match part (non-seed segments). Matches that are found further away from the seed than this value are discarded. Note that in many open source tools users are required to specify the maximum intron size. But here, setting the maximum distance between seed and non seed segments means that this value should be the maximum number of reference bases that a read can span. For long reads spanning many introns, this will be on the order of the length of a typical long gene, and *not* the length of a typical intron.
- Read alignment:
- Match score: The positive score for a match between the read and the reference sequence. It is set by default to 1 but can be adjusted up to 10.
- Mismatch cost: The cost of a mismatch between the read and the reference sequence. Ambiguous nucleotides such as "N", "R" or "Y" in read or reference sequences are treated as mismatches and any column with one of these symbols will therefore be penalized with the mismatch cost.
- Insertion cost: The cost of an insertion in the read (a gap in the reference sequence). cost of an insertion of length l will be l*insertion cost.
- Deletion score: The cost of a deletion in the read (gap in the read sequence). The cost of a deletion of length l will be l*deletion score.
- Length fraction: The minimum percentage of the total alignment length that must match the reference sequence at the selected similarity fraction. A fraction of 0.5 means that at least half of the alignment must match the reference sequence before the full read is included in the mapping (if the similarity fraction is set to 1).
- Similarity fraction: The minimum percentage identity between the aligned region of the read (segment) and the reference sequence. This means that all segments must fulfill this requirement. Since segments can be as short as 15 bp, this threshold should not be set too strictly: for example, setting the threshold at 0.9 means that two errors for a segment of 15 bp would discard the match.
- Non-specific match handling decides whether non-specific matches should be distributed randomly or ignored
Note that in addition to these mapping settings, the Large Gap Read Mapping tool requires that each mapped segment must be of minimum length 15 bp, and that at each mapping step, the mapped segment must comprise at least 10% of the read being mapped. This will initially be the full read length, but in later rounds it will be the length of the remaining unaligned part of the read.
In addition to a reads track (figure 2.4), the tool can generate the following items:
- a report on the mapping, containing various statistics on the mapping, such as the distribution of number of segments per read matching the reference (match parts), the distribution of gaps between the match parts and paired read mapping statistics. In the mapping report, "Unaligned internal gaps" are (small) unmapped parts of the read between mapped segments, whereas "gap between match parts" is the distance between the mapped read segments on the reference. For cDNA reads mapped to genomic sequences, these distances correspond to intron size.
- a list of invalid mapped reads, listing reads for which the Large Gap Read Mapping tool was able to find a mapping, but for which the mappings of the segments were incompatible, i.e., if their positions are not consecutive along the reference, or if they do not have the same direction.
- a list of unmapped reads, for reads that the Large Gap Read Mapping tool was not able to map.
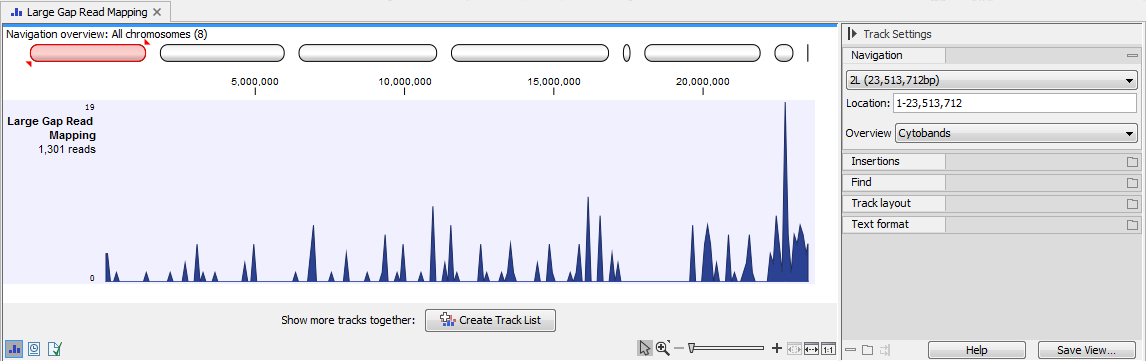
Figure 2.4: The Large Gap Read Mapping track.