Other adapter trimming options
When you run the trim, you specify the adapter settings as shown in figure 23.8.
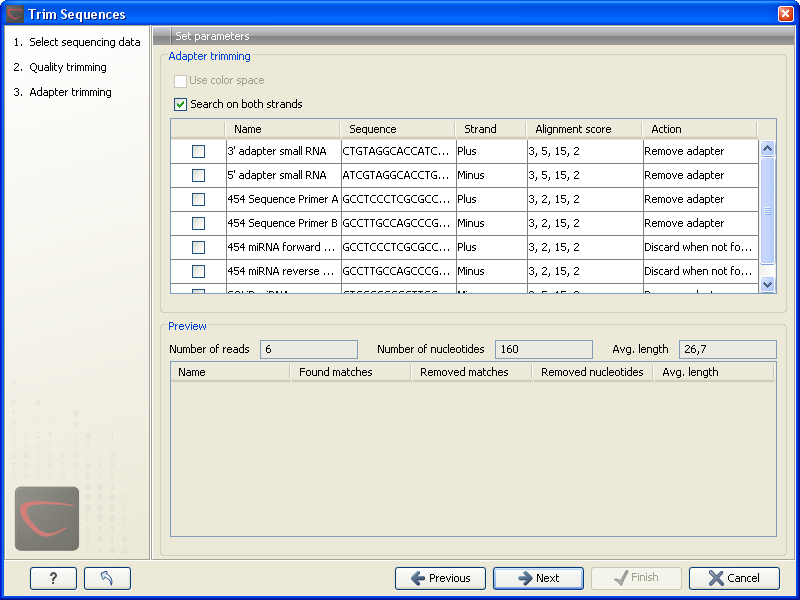
Figure 23.8: Trimming your sequencing data for adapter sequences.
You select an adapter to be used for trimming by checking the checkbox next to the adapter name. You can overwrite the settings defined in the preferences regarding Strand, Alignment score and Action by simply clicking or double-clicking in the table.
At the top, you can specify if the adapter trimming should be performed in Color space. Note that this option is only available for sequencing data imported using the SOLiD import. When doing the trimming in color space, the Smith-Waterman alignment is simply done using colors rather than bases. The adapter sequence is still input in base space, and the Workbench then infers the color codes. Note that the scoring thresholds apply to the color space alignment (this means that a perfect match of 10 bases would get a score of 9 because 10 bases are represented by 9 color residues). Learn more about color space.
Besides defining the Action and Alignment scores, you can also define on which strand the adapter should be found. This can be done in two ways:
- Defining either Plus or Minus for the individual adapter sequence (this can be done either in the Preferences or in the dialog shown in figure 23.8). Note that all the definitions (see here and here) regarding 3' end and 5' end also apply to the minus strand (i.e. selecting the Minus strand is equivalent to reverse complementing all the reads). The adapter in this case should be defined as you would see it on the plus strand of the reverse complemented read. Figure 23.9 below shows a few examples of an adapter defined on the minus strand.
- Checking the Search on both strands checkbox will search both the minus and plus strand for the adapter sequence (the result would be equivalent to defining two adapters and searching one on the plus strand and one on the minus strand).
Below is an example showing hits for an adapter sequence defined as CTGCTGTACGGCCAAGGCG, searching on the minus strand.
|
Below the adapter table you find a preview listing the results of trimming with the current settings on 1000 reads in the input file (reads 1001-2000 when the read file is long enough). This is useful for a quick feedback on how changes in the parameters affect the trimming (rather than having to run the full analysis several times to identify a good parameter set). The following information is shown:
- Name. The name of the adapter.
- Matches found. Number of matches found based on the strand and alignment score settings.
- Reads discarded. This is the number of reads that will be completely discarded. This can either be because they are completely trimmed (when the Action is set to Remove adapter and the match is found at the 3' end of the read), or when the Action is set to Discard when found or Discard when not found.
- Nucleotides removed. The number of nucleotides that are trimmed include both the ones coming from the reads that are discarded and the ones coming from the parts of the reads that are trimmed off.
- Avg. length This is the average length of the reads that are retained (excluding the ones that are discarded).
Next time you run the trimming, your previous settings will automatically be remembered. Note that if you change settings in the Preferences, they may not be updated when running trim because the last settings are always used. Any conflicts are illustrated with text in italics. To make the updated preference take effect, press the Reset to CLC Standard Settings (![]() ) button.
) button.