Downloading reads and metadata from SRA
At the bottom of the SRA results table are two buttons: Download Reads and Metadata and Show Metadata for Selection. The functionality of each is described on this page.
Download Reads and Metadata
Select the rows of interest in the results table and then click on Download Reads and Metadata to download and import reads and metadata from the selected runs. Each resulting sequence list has an association to the row in the CLC Metadata Table with information relevant to those reads. See Finding data elements based on metadata for details about data associations with CLC Metadata Tables.
This CLC Metadata Table can be used directly to define the experiment design for differential expression analyses. CLC Metadata Tables can be edited, if you wish to add or adjust information. See Differential Expression for RNA-Seq and Editing Metadata tables for further details.
Import Options
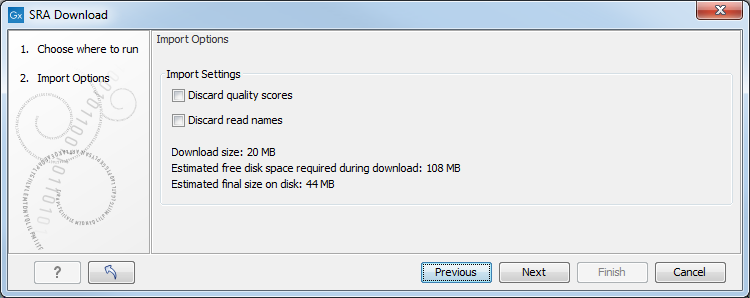
Figure 8.7: Import Options after clicking on Download Reads and Metadata. '
- Discard read names Individual read names are rarely relevant for NGS data. Checking this option can save disk space.
- Discard quality scores Checking this option can reduce disk space usage and memory consumption but should only be done if these scores are not relevant to your work. They are not used for RNA-Seq and expression analyses. They are used during variant detection and can be shown in views of read mappings.
The following information is reported in the wizard:
- Download size The size of the SRA format (.sra) files that will be downloaded. If the SRA file is reference-compressed, a copy of the genome must also be retrieved before the reads can be imported. Thus, the actual download size may be up to 1GB larger than stated in some cases.
- Estimated free disk space required during download A conservative estimate for the disk space required to download the selected runs. This value is the "Estimated final size on disk" + the size of the largest single run in FASTQ format + the size of the largest single run in SRA format.
- Estimated final size on disk An estimate of the total size of the sequence data after import into the Workbench.
See also the section below about how reads are downloaded for more context.
Edit Paired End Settings (figure 8.8)
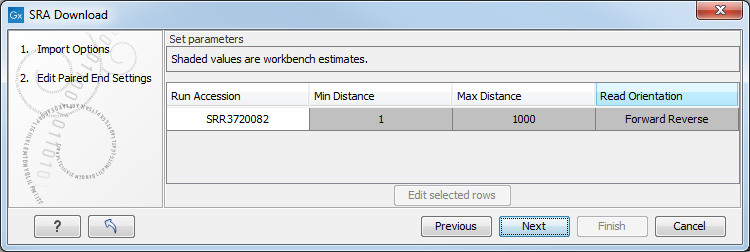
Figure 8.8: The Download Reads and Metadata Edit Paired End Settings dialog.
This dialog appears for all runs marked as being Paired (Paired column contains "Yes").
Read orientation is always guessed to be "Forward Reverse" unless otherwise stated.
Minimum distance and Maximum distance depend on how much data the depositor supplied with the runs. They are allowed to supply an "Insert Size" and an "Insert Deviation".
- If no insert size is supplied, we use defaults of 1 for minimum and 1,000 for maximum.
- If an insert size is supplied, we make the following calculation:
-
- If no deviation is supplied, we estimate this to be
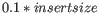 and perform the same calculation as above.
and perform the same calculation as above.
When possible, we generally recommend that SRA data be used in subsequent analyses with the "Auto paired end distance detection" option enabled as the quality of deposited information is low. For example, some depositors report insert size including the length of the reads, and some excluding the length of the reads.