Algorithm details and parameters
The algorithm operates with the following assumption: Mapped reads from duplicated DNA fragments will share a mapping orientation (e.g. will map to the same strand), and depending on their orientation, will share either a start coordinate (forward reads), an end coordinate (reverse reads) or both (paired end reads).
Based on this assumption, a group of reads that share identical start and end coordinates (or start coordinate and length for single end reads) and also share identical sequences can be considered as potential duplications of the same DNA fragment. These reads are then investigated to find reads to be removed and reads to be kept. In order to explain how this works, we will use a small example shown in figure 21.45.
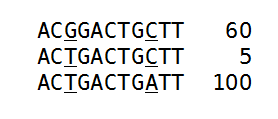
Figure 21.45: An alignment of three different sequence. The numbers are read counts, e.g. the read at the top occurs 60 times.
The example shows 165 reads that share the same start position and orientation and are considered for duplicate read removal. 60 reads share the sequence shown at the top, 5 reads share the middle sequence, and 100 reads share the sequence at the bottom. The differences are at position 3 and 9 (underlined).
The tool will now create a tree structure out of these reads as illustrated in figure 21.46.
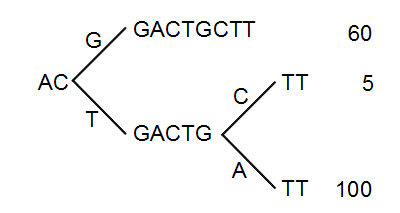
Figure 21.46: The reads from figure 21.45 represented as a Patricia tree[Morrison, 1968].
The first branch point in the tree is at the third position, where sequence number one has a G and the other sequences have a T. The other two sequence disagree at position nine, where one has a C and another has an A.
The next step is to iteratively merge the branches, starting from the end of the tree. The first branch point to consider is at position nine. Since only 5 reads have a C and 100 reads have an A, the C branch is collapsed. This is shown in figure 21.47.
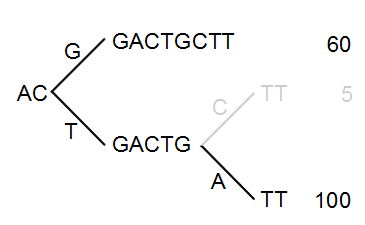
Figure 21.47: Merging the sequences.
As a user, you can specify the threshold for when the reads should be merged. The default is 20 %: when the minority branch has less than 20 % of the read count of the both branches, it is collapsed.
The next branch to consider is at the third position, where there are now 105 reads that have a T and 60 reads that have a G (see figure 21.48).
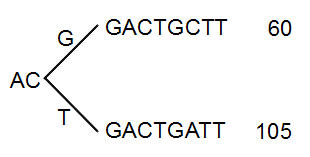
Figure 21.48: Merging the sequences.
With the default setting at 20%, these two branches will not be collapsed, because there are too many reads on the minority branch (60 reads versus 105 reads). Since this process is aimed at collapsing reads that are only distinguished apart by sequencing errors, you would not expect this situation to be caused by sequencing errors, but rather true biological variation (or PCR errors in the early cycles that are indistinguishable from true variation).
If we raised the threshold to 60%, the two branches above would be merged into one if it was not for the second rule governing the merging of branches: The sequences have to be identical except for the difference at the branch point. Looking at the sequences in figure 21.48, there is a difference at position 9 which means that these two branches would never be merged, regardless the threshold and the read counts.
The result of the duplicate reads removal in this example would be that the 165 reads are reduced to two in the result.