Introduction
This user manual is for Whole Genome Alignment 26.0.
The Whole Genome Alignment plugin provides tools for comparing small and medium-sized genomes (up to 100 Mb) and for exploring and visualizing their evolutionary relationships (figure 1.1).
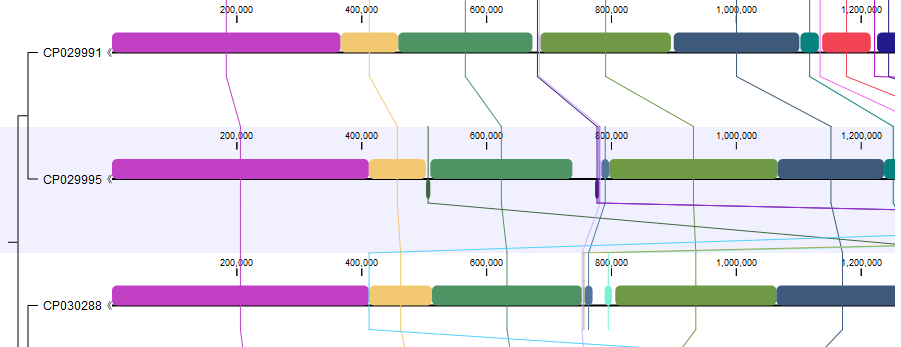
Figure 1.1: A Whole Genome Alignment view.
Tools delivered by this plugin are available under the Whole Genome Alignment folder under the Tools menu (figure 1.2). Relevant importers and exporters are also made available, as described in Import and export.

Figure 1.2: The tools delivered by this plugin are installed in a folder called Whole Genome Alignment, under the Tools menu.
The plugin contains functionality for:
- Generating whole genome dot plots quickly
- Aligning multiple genomes
- Visualizing alignments of multiple genomes, which can contain large-scale events such as inversions and translocations
- Calculating the Average Nucleotide Identity
- Transferring annotations from a reference genome to other genomes
- Creating evolutionary trees and heat maps based on the Average Nucleotide Identity
- Importing and exporting standard Whole Genome Alignments formats (MAF and XMFA)
- Extracting multiple sequence alignments, optionally restricting to annotations such as coding regions
Subsections