The Create SNP Tree tool is inspired by [Kaas et al., 2014].
To generate a SNP tree, first map reads from the individual samples to a common reference and call variants. The corresponding tools are described at:
To create a SNP tree, go to:
Tools | Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | Create SNP Tree (
) | Create SNP Tree (![]() )
)
In the first dialog, select reads tracks or read mappings (figure 9.1).
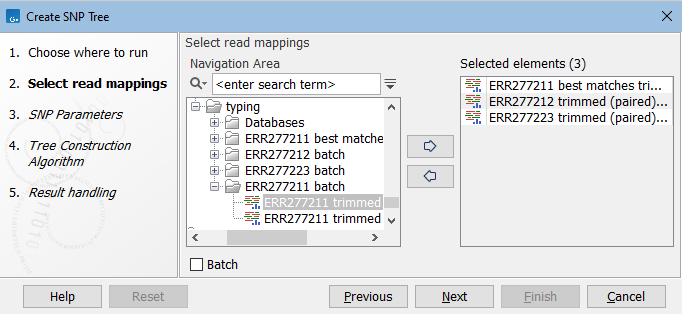
Figure 9.1: Select read mappings to be included in the SNP tree analysis.
Next, select Variant parameters. These determine which SNPs (single-nucleotide polymorphisms) and MNVs (multi-nucleotide variants) to consider for building the SNP tree:
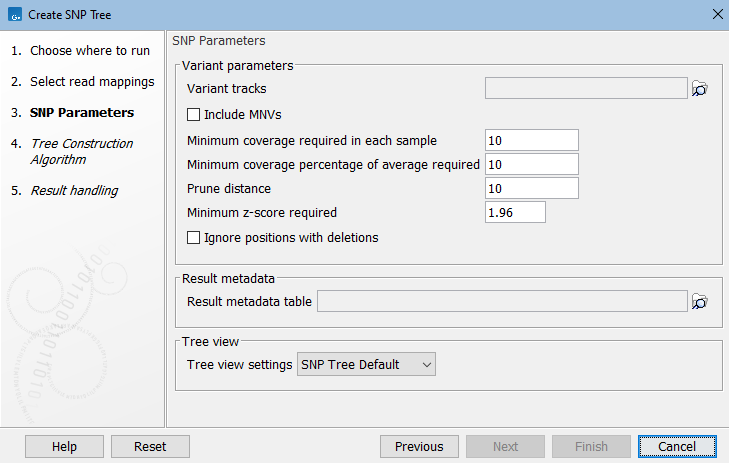
Figure 9.2: Select variant tracks and specify relevant parameters before generation of a SNP tree.
The initial list of SNP positions is reduced based on the above filters. Of the remaining, only variants with relative frequency above 50% (haploid organisms) will be considered. SNP positions that overlap a deletion in any sample are not considered, because such SNPs are often false positives caused by undetected deletions in repeat regions. Information about reference and alleles is deduced from the read mappings.
Optionally, select the Result metadata table with metadata relevant for your samples. This will allow you to decorate the resulting SNP tree with metadata information, see SNP tree.
In the next dialog, select the tree construction algorithm (figure 9.3).
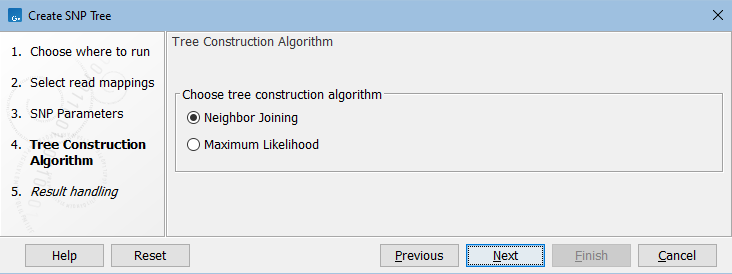
Figure 9.3: Choose the tree construction algorithm.
In rare cases, a SNP position may be determined to be highly unlikely when constructing a Maximum Likelihood tree. When this happens, the position is removed from the SNP alignment and the tree construction is rerun. The number of SNPs removed this way can be seen in the Post-alignment filtering section of the SNP tree report.
If you selected Maximum Likelihood, the next dialog covers parameters for this algorithm (see figure 9.4). The parameters are described here: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Maximum_Likelihood_Phylogeny.html.
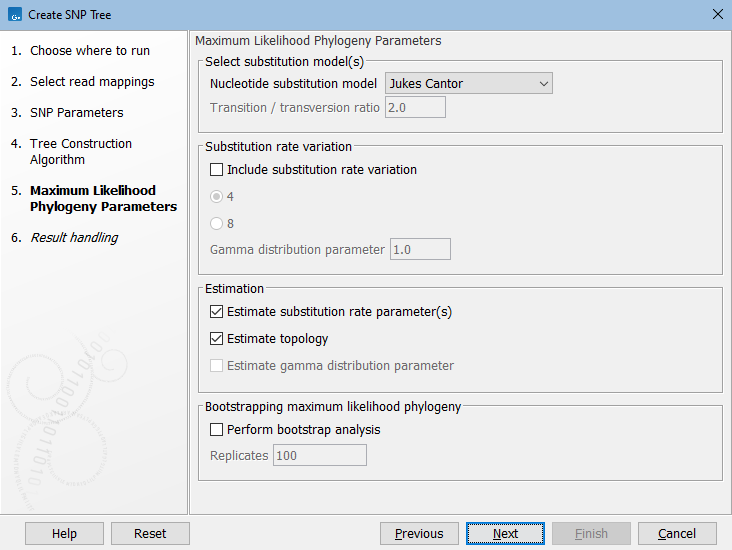
Figure 9.4: Set parameters for maximum likeihood estimation.
In the Result handling dialog, specify the output (figure 9.5).
Figure 9.5: Create SNP Tree output options.
In addition to the SNP tree, the following are available:
At least 100 constant columns are added to ensure that the equilibrium frequencies of nucleotides more closely resemble those in the reference genome. Additional columns are added until the two most distant sequences in the alignment are 80% identical or until the alignment is the size of the reference genome. This partially mitigates the overestimation of branch lengths when using the Maximum Likelihood tree construction algorithm on SNP positions.
The alignment can be used as input for the Model Testing tool that serves to identify which evolutionary model suits the data best. Based on this, you may want to rerun the Create SNP Tree tool with adjusted settings. The Model Testing tool is described at https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Model_Testing.html.