Create Large MLST Scheme
The Create Large MLST Scheme tool can be used to create a scheme from scratch.
To run the Create Large MLST Scheme tool choose:
Microbial Genomics Module (![]() ) | Databases (
) | Databases (![]() ) | Large MLST (
) | Large MLST (![]() ) | Create Large MLST Scheme (
) | Create Large MLST Scheme (![]() )
)
As input, the tool requires a set of complete isolate genomes in the form of one or more sequence lists or sequences. At least one of these genomes must be annotated with coding region (CDS) annotations. If these are not available, the Find Prokaryotic Genes tool (see Find Prokaryotic Genes) or Annotate with DIAMOND (see Annotate with DIAMOND) can be used to predict and annotate the coding regions.
In the first wizard step shown in figure 18.1 the grouping of sequences into genomic units can be controlled. This is necessary when working with genomes that span several chromosomes or several contigs for the tool to consider these as one unit. The grouping can be controlled by the Assembly grouping field:
- Each sequence is one assembly: Each individual sequence is considered a complete assembly of a genome.
- Each input element is one assembly: Each input element, i.e. input sequence or input sequence list, is considered a complete assembly of a genome.
- Group sequences by annotation type: Use annotations to group the assemblies and specify the annotation field with Assembly annotation type. Some tools, such as the Download Microbial Reference Database, will automatically assign an Assembly ID that can be used for grouping. For a manual assignment of Assembly ID annotations, please see Using the Assembly ID annotation.
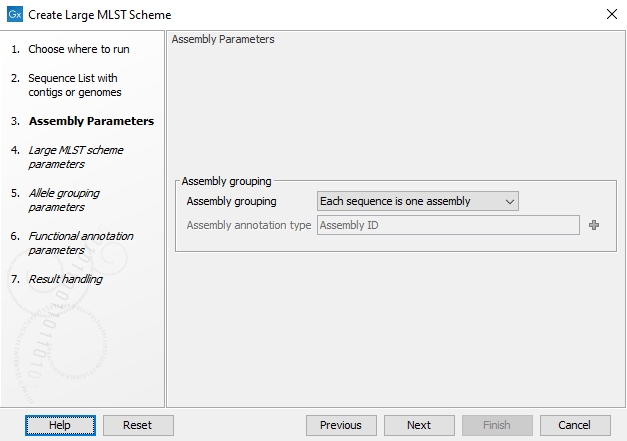
Figure 18.1: Grouping the input into assemblies.
After specifying the input, the second step is to set up the basic Large MLST Scheme creation parameters (figure 18.2).
The Create Large MLST Scheme tool works by extracting all annotated coding sequences (CDS) and clustering them into similar gene classes (loci). It is possible to specify whether we are interested in the genes that are present in some genomes (Whole genome - must be present in at least 10% of all genomes), most genomes (Core genome - must be present in at least 90% of the genomes), or a user-specified Minimum fraction.
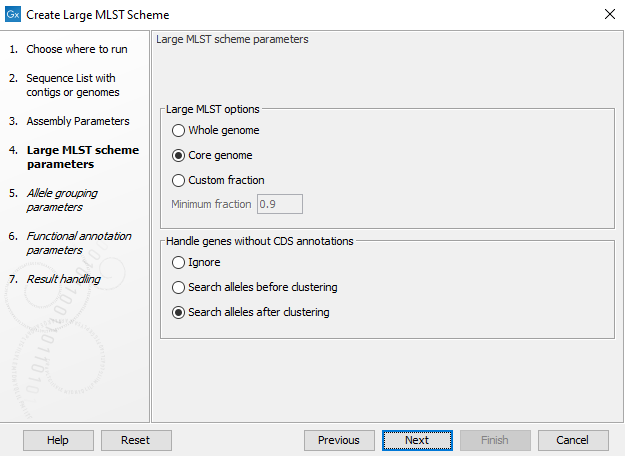
Figure 18.2: Basic options for creating a large MLST scheme.
The best results are obtained by supplying genomes with proper CDS annotations. The Handle genes without annotations option controls how genomes without CDS annotations and how existing CDS may be overridden if a longer CDS from another genome exactly matches the genomic sequence.
- Ignore: Only use the existing CDS annotations as a basis for the large MLST scheme construction.
- Search alleles before clustering: All of the input genomes are blasted (using DIAMOND) against the set of annotated genes, and any new genes will be added as alleles. This is a very slow, but thorough check.
- Search alleles after clustering: After clustering the genes, all of the input genomes are blasted (using DIAMOND), but only against the longest protein in each cluster.
The Allele grouping parameters step (figure 18.3) specifies how the different genes (CDS annotations) are compared to each other. DIAMOND is used for this clustering. The following can be specified:
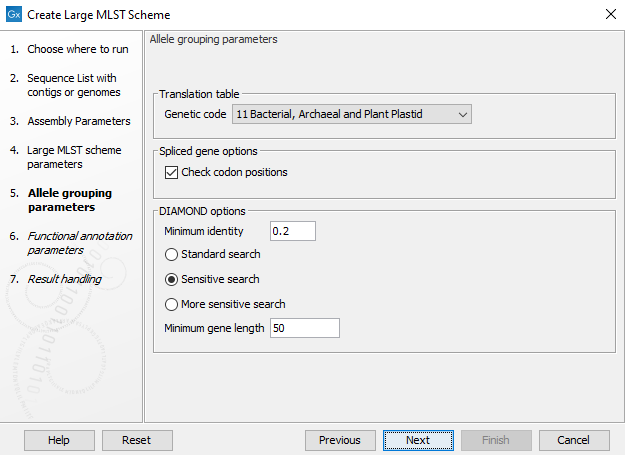
Figure 18.3: The allele grouping (clustering) options.
- Genetic code: specifies the genomic code to use for the input samples if Check codon positions is enabled.
- Check codon positions: If this is enabled, coding sequences not starting with a start codon, not ending with a stop codon or containing internal stop codons will be discarded. This can be disabled, for example to allow the construction of MLST schemes with spliced genes where each exon is considered an allele.
- Minimum identity: determines the minimum sequence identity before grouping protein sequences.
- It is also possible to specify the sensitivity of the search (Standard search, Sensitive search, and More sensitive search) - increasing the sensitivity makes the search more thorough, but also much slower. The default for this parameter is Sensitive search.
- Minimum gene length: is used to remove short genes from the resulting large MLST scheme.
It is possible to decorate the alleles with information about virulence or resistance. The information can be extracted from either a ShortBRED Marker database or a Nucleotide database. These databases can be accessed using the Download Resistance Database tool (e.g. QMI-AR for resistance or VFDB for virulence) and can be provided as input to the Create Large MLST Scheme tool at this step (figure 18.4).
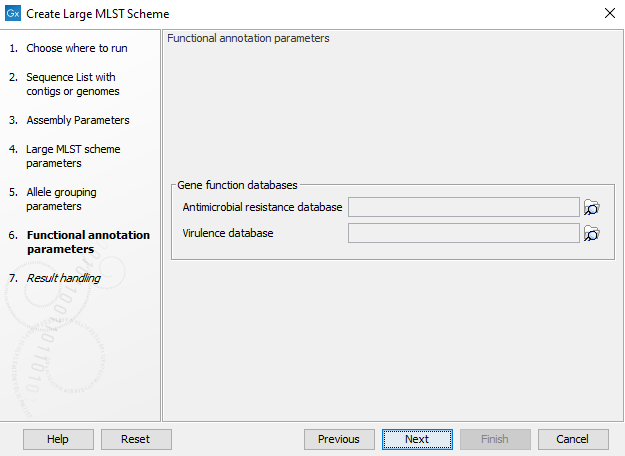
Figure 18.4: The functional annotation parameters.