Annotate tag experiment
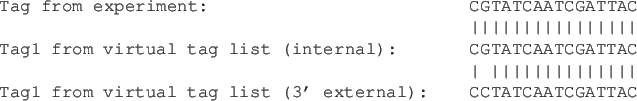
Combining the tag counts ( ) from the experimental data (see Extract and count tags) with the virtual tag list (
) from the experimental data (see Extract and count tags) with the virtual tag list ( ) (see Create virtual tag list) makes it possible to put gene or transcript names on the tag counts. The Workbench simply compares the tags in the experimental data with the virtual tags and transfers the annotations from the virtual tag list to the experimental data.
) (see Create virtual tag list) makes it possible to put gene or transcript names on the tag counts. The Workbench simply compares the tags in the experimental data with the virtual tags and transfers the annotations from the virtual tag list to the experimental data.
This is done on an experiment level (experiments are collections of samples with defined groupings, see Experimental design):
Toolbox | Transcriptomics Analysis ( ) | Expression Profiling by Tags (
) | Expression Profiling by Tags ( ) | Annotate Tag Experiment (
) | Annotate Tag Experiment ( )
)
You can also access this functionality at the bottom of the Experiment table ( ) as shown in figure 28.43.
) as shown in figure 28.43.
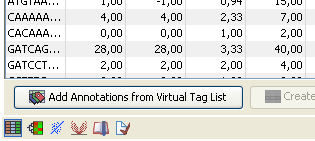
Figure 28.43: You can annotate an experiment directly from the experiment table.
This will open a dialog where you select a virtual tag list () and an experiment () of tag-based samples. Click Next when the elements are listed in the right-hand side of the dialog.
This dialog lets you choose how you want to annotate your experiment (see figure 28.44).
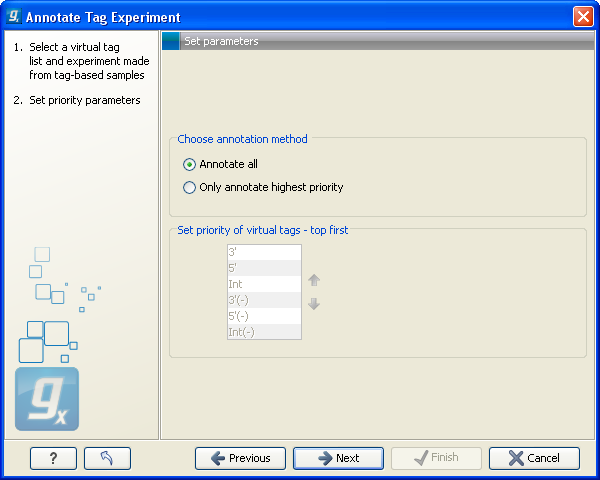
Figure 28.44: Defining the annotation method.
If a tag in the virtual tag list has more than one origin (as shown in the example in figure 28.42) you can decide how you want your experimental data to be annotated. There are basically two options:
- Annotate all
- This will transfer all annotations from the virtual tag. The type of origin is still preserved so that you can see if it is a 3' external, 5' external or internal tag.
- Only annotate highest priority
- This will look for the highest priority annotation and only add this to the experiment. This means that if you have a virtual tag with a 3' external and an internal tag, only the 3' external tag will be annotated (using the default prioritization). You can define the prioritization yourself in the table below: simply select a type and press the up (
 ) and down (
) and down ( ) arrows to move it up and down in the list. Note that the priority table is only active when you have selected Only annotate highest priority.
) arrows to move it up and down in the list. Note that the priority table is only active when you have selected Only annotate highest priority.
Click Next to choose how you want to tags to be aligned (see figure 28.45).
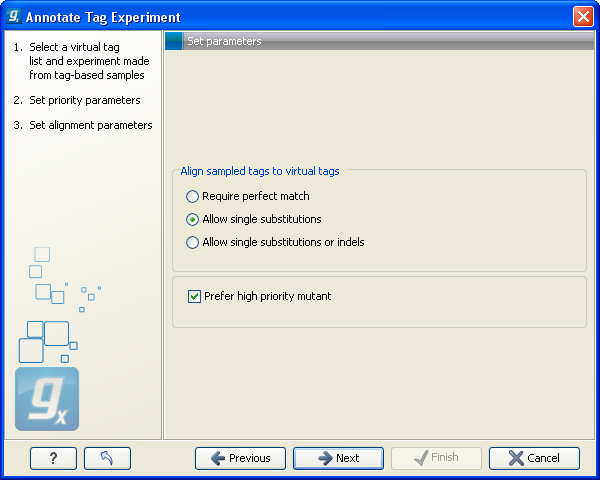
Figure 28.45: Settings for aligning the tags.
When the tags from the virtual tag list are compared to your experiment, the tags are matched using one of the following options:
- Require perfect match
- The tags need to be identical to be matched.
- Allow single substitutions
- If there is up to one mismatch in the alignment, the tags will still be matched. If there is a perfect match, single substitutions will not be considered.
- Allow single substitutions or indels
- Similar to the previous option, but now single-base insertions and deletions are also allowed. Perfect matches are preferred to single-base substitutions which are preferred to insertions, which are again preferred to deletions. 28.7
If you select any of the two options allowing mismatches or mismatches and indels, you can also choose to Prefer high priority mutant. This option is only available if you have chosen to annotate highest priority only in the previous step (see figure 28.44). The option is best explained through an example:
In this case, you have a tag that matches perfectly to an internal tag from the virtual tag list. Imagine that in this example, you have prioritized the annotation so that 3' external tags are of higher priority than internal tags. The question is now if you want to accept the perfect match (of a low priority virtual tag) or the high-priority virtual tag with one mismatch? If you check the Prefer high priority mutant, the 3' external tag in the example above will be used rather than the perfect match.
Click Next if you wish to adjust how to
handle the results. If not, click Finish.
. This will add extra annotation columns to the experiment. The extra columns corresponds to the columns found in your virtual tag list. If you have chosen to annotate highest priority-only, there will only be information from one origin-column for each tag as shown in figure 28.46.
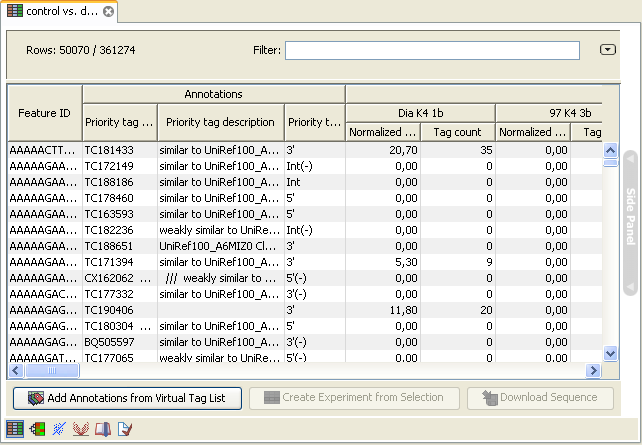
Figure 28.46: An experiment annotated with prioritized tags.
The CLC Genomics Workbench is able to analyze expression data produced on microarray platforms and high-throughput sequencing platforms (also known as Next-Generation Sequencing platforms).
The CLC Genomics Workbench provides tools for performing quality control of the data, transformation and normalization, statistical analysis to measure differential expression and annotation-based tests. A number of visualization tools such as volcano plots, MA plots, scatter plots, box plots, and heat maps are used to aid the interpretation of the results.
Footnotes
- ... deletions.28.7
- Note that if you use color space data, only color errors are allowed when choosing anything but perfect match.