PacBio De Novo Assembly Pipeline
Please note that the tools Correct PacBio Reads and De Novo Assemble PacBio Reads are optimized for the use of PacBio data and readily support data generated with different generations of PacBio chemistry (sequencing reagents). Due to such algorithm-optimizations the use of these tools for other data types is not supported. Moreover, for the tool Correct PacBio Reads we are relying on certain methods which are the intellectual property of Pacific Biosciences. The use of the Correct PacBio Reads tool or the predefined workflow PacBio De Novo Assembly Pipeline with any data other than data generated on a Pacific Biosciences instrument constitutes a violation of the end user license agreement that users of the CLC Genome Finishing Module agree to during installation.
The PacBio De Novo Assembly Pipeline workflow (see figure 18.1) takes PacBio reads that have already been imported (see Import PacBio Reads) as input and produces a high-quality assembly together with a number of reports that can be used to evaluate the quality of both the input data and the assembly. It consists of seven steps running six different tools from the CLC Genome Finishing toolbox and the general CLC Genomics Workbench toolbox:
- Raw PacBio reads import Raw PacBio reads are imported from FASTQ or H5 files (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Import_high_throughput_sequencing_data.html.
- Correct PacBio Reads Sequencing errors are corrected and chimeric reads and untrimmed adapters are resolved in a subset of the longest reads in the input data set. The corrected reads are output in a file named 'Corrected reads' and a summary of the error-correction is saved in a file named 'Corrected reads - report'. This report can be used to both evaluate the quality of the input reads and to assess the error-correction and assembly parameters.
- De Novo Assemble PacBio Reads The error-corrected reads are assembled into high-quality contigs.
- Map Reads to Contigs The corrected reads are mapped to the contigs in order to be able to run the Join Contigs tool.
- Join Contigs Contigs are joined by automatic scaffolding based on the read mapping created above. The final contigs are saved to a file named 'Contig sequences'.
- Map Reads to Contigs The corrected reads are mapped to the final contigs in order to be able to run the Analyze Contigs tool. This read mapping can, together with the output from the Analyze Contigs tool, furthermore be used to evaluate the support for each contig and manually identify and resolve possible assembly errors. The read mapping is saved to a file named 'Corrected reads mapped to contigs' and a report that summarizes the read mapping is saved to a file named 'Corrected reads mapped to contigs - report'.
- Analyze Contigs The final contigs are analyzed in order to find problematic regions that may need manual curation. A summary of the analysis is saved to a file named 'Contig analysis report' and the problematic regions are reported in a file named 'Contig analysis table'.
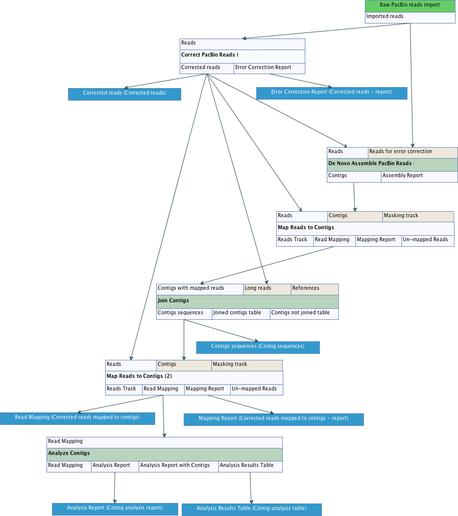
Figure 18.1: The PacBio De Novo Assembly Pipeline workflow.