LightSpeed Fastq to Somatic Variants Tumor Normal
The LightSpeed Fastq to Somatic Variants Tumor Normal tool is designed to provide somatic variant calls from a tumor and a normal sample within a very short timeframe.
The tool can perform read trimming, mapping, deduplication, local realignment and variant calling. For a description of each step, see LightSpeed Methods.
LightSpeed Fastq to Somatic Variants Tumor Normal can only analyze one sample per analysis start.
To run the tumor normal LightSpeed tool go to:
Tools | LightSpeed (![]() ) | LightSpeed Fastq to Somatic Variants Tumor Normal (
) | LightSpeed Fastq to Somatic Variants Tumor Normal (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the first wizard step, specify tumor and normal fastq files and a reference sequence (figure 3.11):
- Input data
- Tumor reads (fastq) Tumor fastq files for analysis. At least two fastq files representing R1 and R2 reads must be provided.
- Normal reads (fastq) Normal fastq files for analysis. At least two fastq files representing R1 and R2 reads must be provided.
- References
- References The reference sequence that reads will be mapped to.
- Reference masking
- No masking Reads are mapped to the full reference sequence.
- Exclude annotated Reads are mapped to the full reference sequence except regions specified in the masking track.
- Include annotated only Reads are only mapped to the regions specified in the masking track.
- Masking track The track specifying the masking regions.
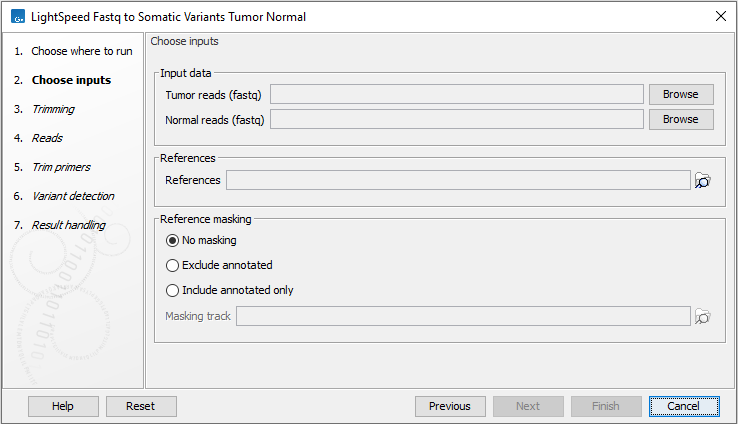
Figure 3.11: Input fastq files and references, and, optionally, a track for reference masking.
Next, options are available for trimming (figure 3.12):
- Trimming
- Quality trim Reads are trimmed for low quality nucleotides.
- Quality trim limit Adjust the quality trim limit for softer or harder trimming. Read more about the quality trim limit here: http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Quality_trimming.html.
- Minimum read length after quality trim Trimmed reads shorter than this length are removed.
- Adapter trim Reads are trimmed for read-through adapter sequence.
- Minimum read length after adapter trim Trimmed reads shorter than this length are removed.
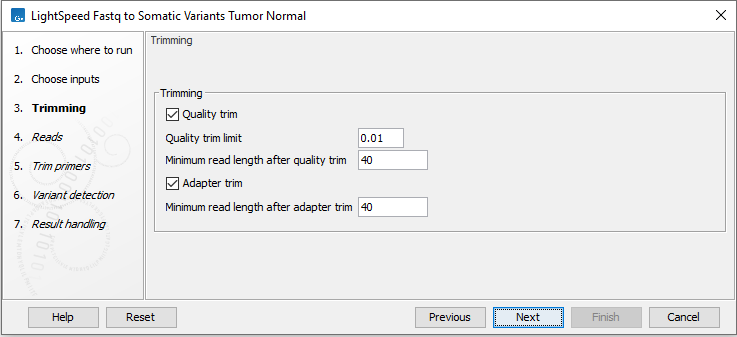
Figure 3.12: Options for trimming.
Next, options are available for UMI and duplicate reads (figure 3.13):
- UMI
- UMI preset Set UMI status of reads.
- No UMI Select for reads that do not have UMIs.
- Custom Select to specify a protocol-specific read and UMI structure.
- QIAseq Targeted DNA Select if you are analyzing data from a QIAseq Targeted DNA panel. The UMI and common sequence position and length are automatically adjusted to match the panel design.
- QIAseq Targeted DNA Pro Select if you are analyzing data from a QIAseq Targeted DNA Pro panel. The UMI and common sequence position and length are automatically adjusted to match the panel design.
- UMI length (Read 1) The number of nucleotides at the start of read 1 that are part of the UMI.
- Common sequence length (Read 1) The number of nucleotides between the UMI and the biological sequence in read 1. These nucleotides are discarded.
- UMI length (Read 2) The number of nucleotides at the start of read 2 that are part of the UMI.
- Common sequence length (Read 2) The number of nucleotides between the UMI and the biological sequence in read 2. These nucleotides are discarded.
- Minimum UMI group size Only UMI groups consisting of at least this number of input read pairs will be merged to consensus UMI reads. UMI groups with fewer input read pairs than this number will be discarded. A UMI group is a group of input read pairs with the same UMI sequence that maps to the same genomic position.
- Maximum UMI differences Add input read pairs to the same UMI group when their UMI sequences have at most this number of differences. One difference is a single nucleotide mismatch, insertion, or deletion.
- UMI window size Add input read pairs to the same UMI group when they map to genomic positions at most this number of bases apart.
- UMI preset Set UMI status of reads.
- Mapped read handling
- Discard duplicate mapped reads Reads likely representing PCR duplicates are collapsed. This option is disabled when UMIs are used to group reads.
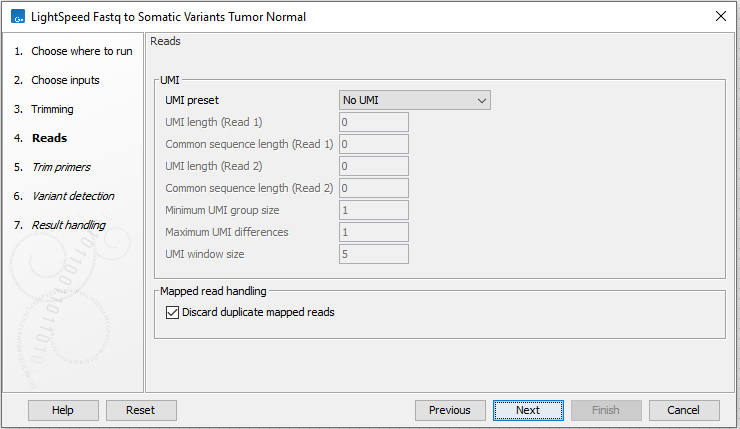
Figure 3.13: Options for UMI and duplicate reads.
Next, options are available for primer trimming (figure 3.14):
- Trim primers
- No primer trim Disable the trim primers step.
- Start of read 1 Primers are at the start of read 1.
- Start of read 2 Primers are at the start of read 2.
- Primers track Annotation track with location and strand of primers. Unalign parts of mapped reads that overlap a primer.
- Discard reads without primer Discard reads that do not overlap with a primer in the primers track.
- Minimum read length after primer trim Reads that are shorter than this number of nucleotides after primer trim are discarded.
- Additional bases to trim Unalign this number of additional mapped bases in reads matching a primer. Bases are unaligned at the beginning of the mapped read downstream from the primer.
- Minimum primer overlap (%) Reads overlap a primer when the expected part of the read (start of read 1 or read 2) maps to a genomic location that overlaps at least this percentage of a primer. Reads that do not meet the threshold are discarded.
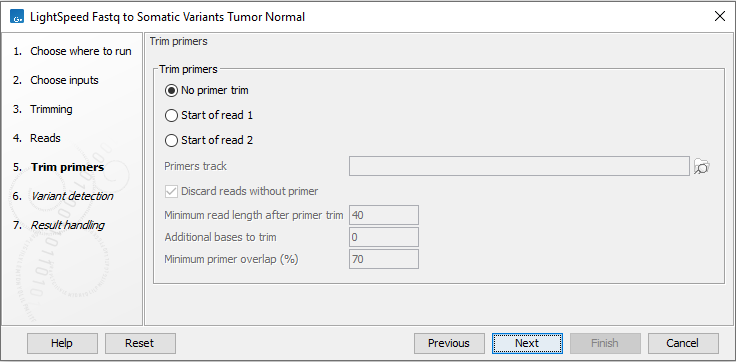
Figure 3.14: Options for primer trimming.
A number of options are available for variant detection (figure 3.15).
- Variant detection
- Restrict calling to target regions Optional. A track defining where variants are called.
- Ignore non-specific matches Reads that map equally well to more than one genomic position, are not used for variant calling.
- SNV minimum allele count Minimum allele count required for SNVs and MNVs to be called.
- SNV minimum per-strand allele count Minimum allele count required on each strand for SNVs and MNVs to be called.
- Indel minimum allele count Minimum allele count required for indels to be called.
- Indel minimum per-strand allele count Minimum allele count required on each strand for indels to be called.
- SNV significance threshold using global error rate p-value threshold for SNVs and MNVs. The p-value is calculated from a binomial test given count, coverage and an error rate of 0.005. Allowed range: 0 - 0.1.
- Indel significance threshold using global error rate p-value threshold for indels. The p-value is calculated from a binomial test given count, coverage and an error rate of 0.005. Allowed range: 0 - 0.1.
- Significance threshold using local error rate p-value threshold for all variants. The p-value is the minimum p-value from two individual tests: 1. A binomial test given forward count, forward coverage and a local error rate for forward reads estimated from the data. 2. A binomial test given reverse count, reverse coverage and a local error rate for reverse reads estimated from the data. Allowed range: 0 - 0.1.
- Significance threshold for low complexity regions p-value threshold for variants that are located in positions where two upstream and two downstream reference symbols are identical to the variant. The p-value is calculated from a binomial test given count and coverage. This filter only removes alleles that have a frequency greater than 10(%). Allowed range: 0 - 1.
- SNV strand balance threshold Threshold for a strand balance score for SNVs and MNVs. The score is calculated as 1 - (p-value from binomial test given forward count, count, and forward count/coverage). Allowed range: 0.9 - 1.
- Indel strand balance threshold Threshold for a strand balance score for indels. The score is calculated as 1 - (p-value from binomial test given forward count, count, and forward count/coverage). Allowed range: 0.9 - 1.
- Normal filters
- Maximum count in normal Somatic variants are not reported when the variant count in the normal is equal to or higher than this threshold.
- Maximum frequency in normal (%) Somatic variants are not reported when the variant frequency in the normal is equal to or higher than this threshold.
- Significance threshold using global error rate in normal Somatic variants are not reported when the variant p-value in the normal is lower than this threshold. Allowed range: 0 - 0.1.
For the options under Variant detection
- All of the options, except "SNV minimum allele count" and "Indel minimum allele count" only removes alleles with a frequency of less than 30(%).
- For all of the options that include threshold in the name, lowering the value will reduce the number of called variants.
Figure 3.15: Options for variant detection.
In the final wizard step, choose which outputs should be generated and whether results should be saved or opened. If a reads track is selected as output, runtime will increase.
Subsections