Demultiplex reads
Multiplexing techniques are often used when sequencing different samples in one sequencing run. One method used is to tag the sequences with a unique identifier during the preparation of the sample for sequencing [Meyer et al., 2007].
With this technique, each sequence read will have a sample-specific tag, which is a specific sequence of nucleotides before and after the sequence of interest. This principle is shown in figure 19.16.
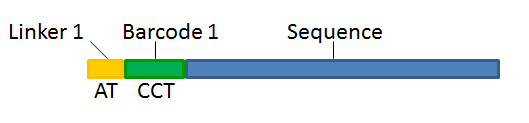
Figure 19.16: Tagging the target sequence, which in this case is single reads from one sample.
The sample-specific tag, also called the barcode or the index, can then be used to distinguish between the different samples when analyzing the sequencing data.
Post-processing of the sequencing data is required to separate the reads into their corresponding samples. Based on their barcodes this can be done using the demultiplexing functionality of the Biomedical Genomics Workbench. Using this tool, sequences are associated with a particular sample when they contain an exact match to a particular barcode. Sequences that do not contain an exact match to any of the barcode sequences provided are classified as not grouped and are put into a sequence list with the name "Not grouped".
Note that there is also an example using Illumina data here.
Before processing the data, you need to import it as described in Import high-throughput sequencing data.
Please note that demultiplexing is often carried out on the sequencing machine so that the sequencing reads are already separated according to sample. This is often the best option, if it is available to you. Of course, in such cases, the data will not need to be demuliplexed again after import into the Biomedical Genomics Workbench.
To demultiplex your data, please go to:
Toolbox | Preparing Raw Data (![]() ) | Demultiplex Reads (
) | Demultiplex Reads (![]() )
)
This opens a dialog where you can specify the sequences to process (figure 19.17).
Figure 19.17: Specify the sequences to demultiplex.
When you click on the button labeled Next, you can then specify the details of how the demultiplexing should be performed. At the bottom of the dialog, there are three buttons, which are used to Add, Edit, and Delete the elements that describe how the barcode is embedded in the sequences.
First, click Add to define the first element. This will bring up the dialog shown in 19.18.
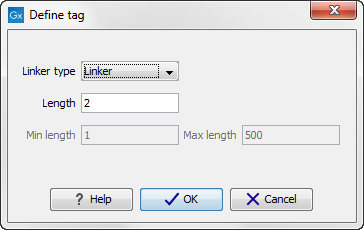
Figure 19.18: Defining an element of the barcode system.
At the top of the dialog, you can choose the type of element you wish to define:
- Linker. The linker (also known as adapter) is a sequence which should just be ignored - it is neither the barcode nor the sequence of interest. In the example in figure 19.16, the linker is two nucleotides long. For this element, you simply define its length - nothing else.
- Barcode. The barcode (also known as index) is the stretch of nucleotides used to group the sequences. In this dialog, you simply need to specify the length of the barcode. The valid sequences for your barcodes must be provided at a later wizard step.
- Sequence. This element defines the sequence of interest. You can define a length interval for how long you expect this sequence to be. The sequence part is the only part of the read that is retained in the output. Both barcodes and linkers are removed.
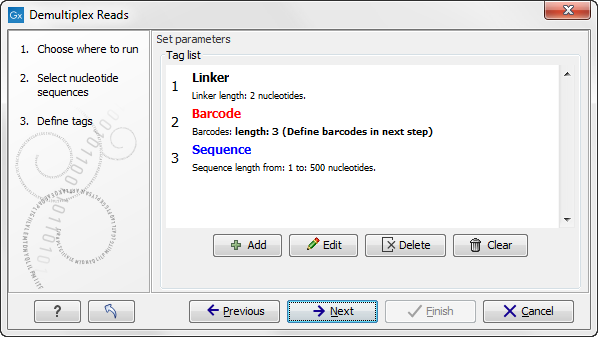
Figure 19.19: Processing the tags as shown in the example of figure 19.16.
Click on the button labeled Next to go to the last wizard step, where you can specify the output options. If you choose to keep the default settings, three different types of output will be generated; 1) The demultiplexed reads, one output for each specified barcode (the file name starts with "Barcode:" and is followed by the specified barcode sequence), 2) The discarded reads that did not have a barcode (this file is called "Not grouped"), and 3) a "Demultiplex Reads report", which shows the fraction of reads with and without a barcode (see figure 19.20).
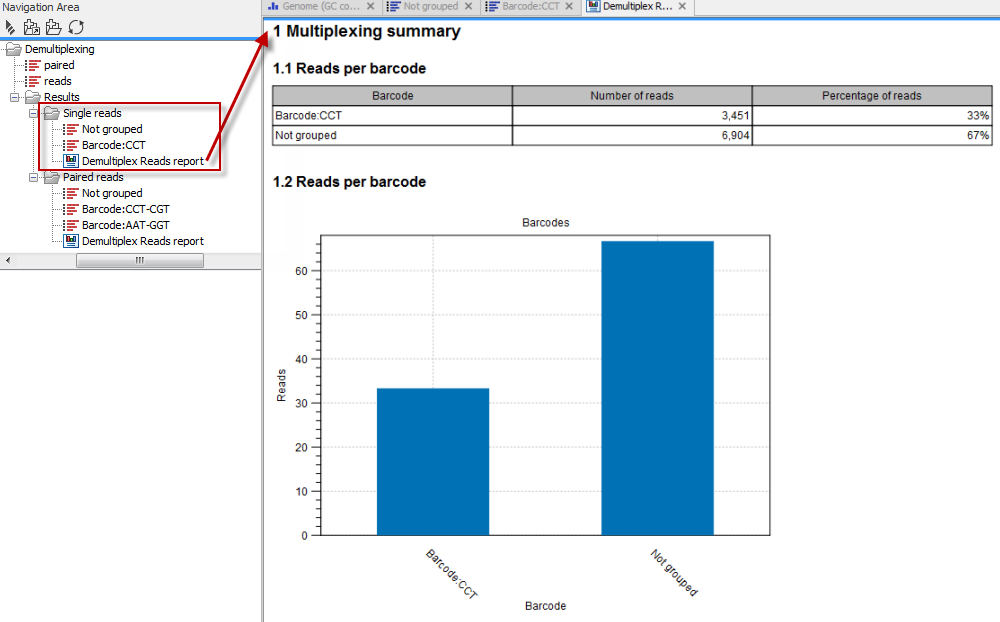
Figure 19.20: Three different outputs are generated when analyzing single reads with only one sample using the default output settings. If several samples had been mixed together there would be a sequence list for each sample (each specified barcode). The Demultiplex Reads report is shown in the right-hand side of the figure.
If you have paired data the procedure is exactly the same, except for one thing: the dialog shown in figure 19.19 will be displayed twice - one for each part of the pair.
Figure 19.21 shows an example that could illustrate paired end reads. In this example we have paired end reads from two different samples mixed together.
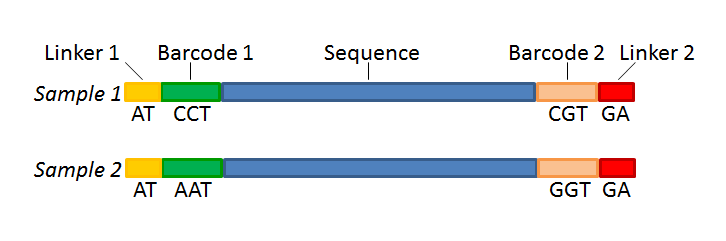
Figure 19.21: Paired end reads with linkers and barcodes. Two different samples are mixed together in this example.
In a situation where the paired reads are expected to be barcoded in the same way (see example below), you would set the parameters for read1 (wizard step 3) and read2 (wizard step 4) to be the same.
Read1 : -Linker-Barcode1-Sequence
Read2 : -Linker-Barcode1-Sequence
However, if read2 of the pair is not expected to be the same as read1 in the pair, it is necessary to adjust these settings accordingly. For example, it is possible that read2 does not contain any barcode sequence at all. In this case, you would simply set the sequence parameter for the mate and exclude the barcode and linker parameters. If the two reads in the read pair have different barcodes, the situation would look like this:
Read1 : -Linker-Barcode1-Sequence
Read2 : -Linker-Barcode2-Sequence
To demultiplex paired end reads the first two steps are similar to the demultiplexing of single reads. Select the paired end read sequences that should be demultiplexed (figure 19.22).
Figure 19.22: Specifying the paired end reads to demultiplex.
Click on the button labeled Next to define the tags and sequence for the forward reads (figure 19.23).
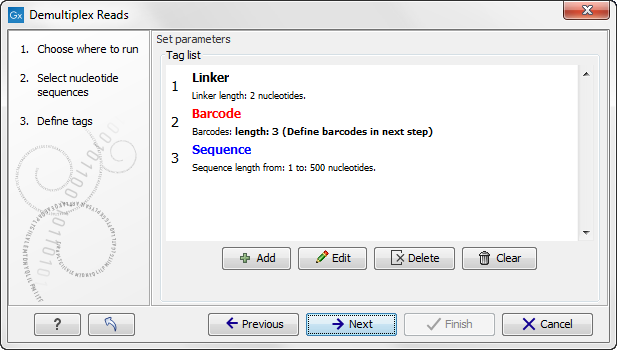
Figure 19.23: Specifying the setup of the forward read (tags and sequence).
Barcodes can be entered manually or imported from a properly formatted CSV or Excel file:
Manually The barcodes can be entered manually by clicking the Add (![]() ) button. In the example shown in figure 19.21 the barcodes should be defined as shown in figure 19.24.
) button. In the example shown in figure 19.21 the barcodes should be defined as shown in figure 19.24.
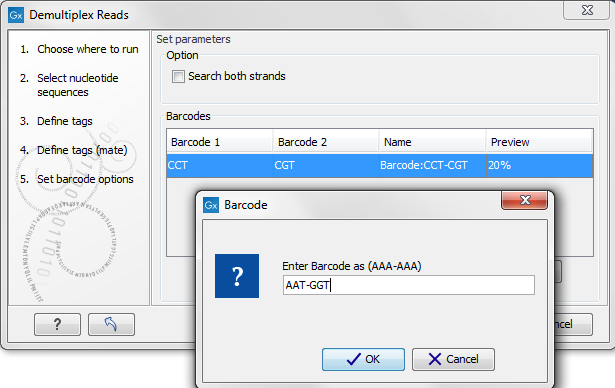
Figure 19.24: The barcodes for the set of paired end reads for sample 1 have already been defined and the barcodes for sample 2 is being entered in the format AAA-AAA, which corresponds to Barcode1-Barcode2 for sample 2 in the example shown in figure 19.21.
You can edit the barcodes and the names by clicking the cells in the table. The barcode name is used when naming the results.
Import from CSV or Excel To import a file of barcodes, click on the Import (![]() ) button.
The input format consists of two columns: the first contains the barcode sequence, the second contains the name of the barcode. An acceptable csv format file would contain columns of information that looks like:
) button.
The input format consists of two columns: the first contains the barcode sequence, the second contains the name of the barcode. An acceptable csv format file would contain columns of information that looks like:
| "AAAAAA","Sample1" |
| "GGGGGG","Sample2" |
| "CCCCCC","Sample3" |
Note that double quotes around values are always necessary regardless of whether columns are separated with a comma or a semi-colon.
The Preview column will show a preview of the results by running through the first 10,000 reads (figure 19.25).
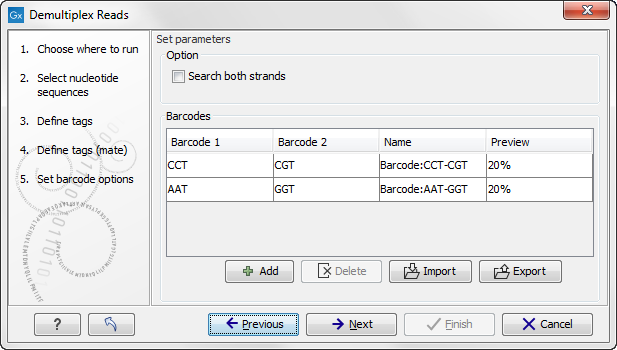
Figure 19.25: A preview of the results.
At the top, you can choose to search on both strands for the barcodes (this is needed for some 454 protocols where the MID is located at either end of the read).
If you would like to change the name of the sequence(s), this can be done at this step by double-clicking on the specific name that you would like to change. This is shown in figure 19.26.
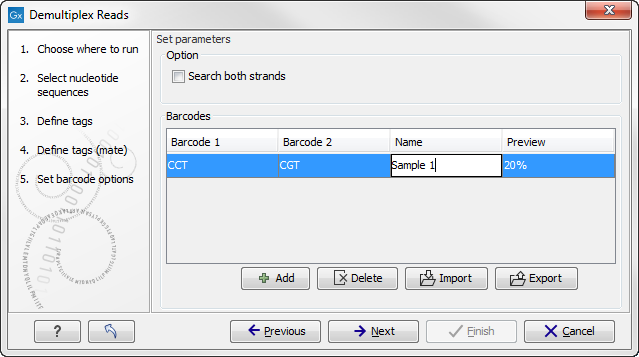
Figure 19.26: The name of the sequence can be renamed by double-clicking on the existing name.
Click on the button labeled Next to define the tags and sequence for the reverse reads (figure 19.27).
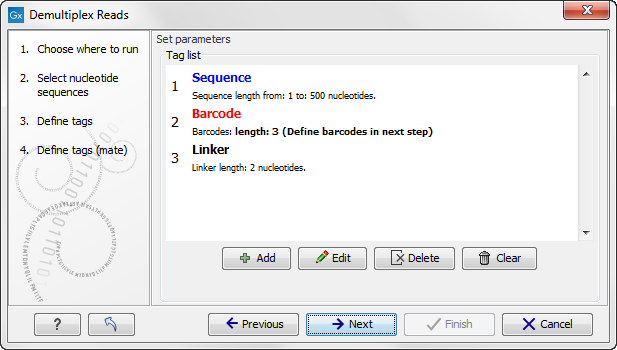
Figure 19.27: Specifying the setup of the reverse read (tags and sequence).
Click Next to specify the output options. First, you can choose to create a list of the reads that could not be grouped. Second, you can create a summary report showing how many reads were found for each barcode. Click Finish to perform the demultiplexing.
There is also an option to create subfolders for each sequence list. This can be handy when the results need to be processed in batch mode.
A new sequence list will be generated for each barcode containing all the sequences where this barcode is identified. Both the linker and barcode sequences are removed from each of the sequences in the list, so that only the target sequence remains. This means that you can continue the analysis by doing trimming or mapping. Note that you have to perform separate mappings for each sequence list.
An example of the demultiplexing summary report is shown in figure 19.28.
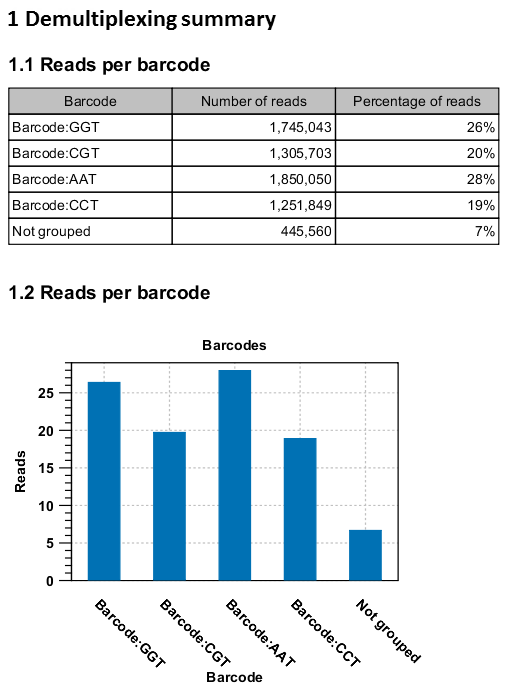
Figure 19.28: An example of a report showing the number of reads in each group. In this example four different barcodes were used to separate four different samples.