Advanced Structural Variant Detection
To run the Advanced Structural Variant Detection (beta) tool:
Toolbox | Resequencing Analysis (![]() ) | Variant Detection (
) | Variant Detection (![]() ) | Advanced Structural Variant Detection (
) | Advanced Structural Variant Detection (![]() )
)
Once the tool wizard has opened (figure 2.1), choose the input you would like to use. The Advanced Structural Variant Detection tool accepts read mappings as either reads tracks or stand-alone read mappings.
Figure 2.1: Select one or several reads tracks or stand-alone read mappings.
The tool has the following options (figure 2.2):
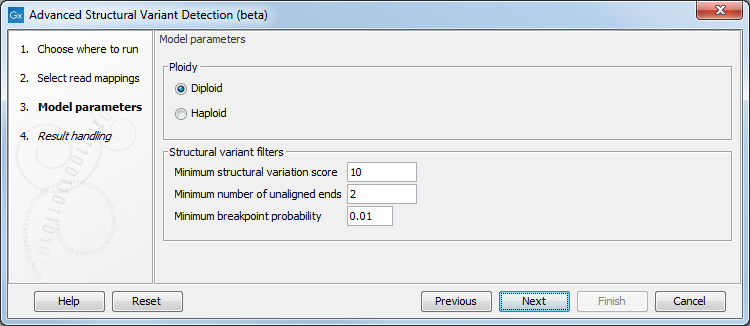
Figure 2.2: Set the model parameters for the tool.
- Ploidy Determines the ploidy of the sample. The value will be used in the statistical model that assign probabilities to breakpoints.
- Minimum structural variation score Measure of how likely this structural variant is. This value may be increased to reduce the number of structural variants called.
- Minimum number of unaligned ends Minimum number of reads with unaligned ends required to detect a breakpoint.
- Minimum breakpoint probability Probability of a breakpoint is based on a statistical model.
The tool outputs a report, a Breakpoint track (BP), and 2 Variant track (SV) and (SV annotations)
The report
The report (figure 2.3) gives an overview of the numbers and types of structural variants found in the sample.
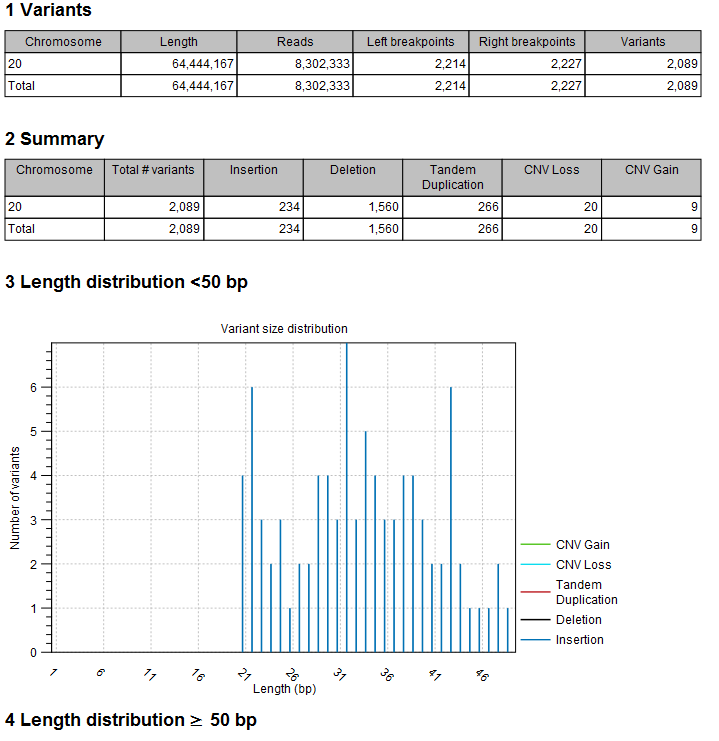
Figure 2.3: The Advanced Structural Variant Detection (beta) report.
It contains:
- A 'Variants' table with a row for each reference sequence, and information on the number of left and right unaligned breakpoint signatures and the resulting number of structural variants found.
- A 'Summary' table with a row for each reference sequence, and information on the total number of variants, stratified into the different variant categories (Insertion, Deletion, Tandem Duplication, CNV Loss, CNV Gain).
- A length distribution plot for short (<50 bp) structural variants
- A length distribution plot for long (>50 bp) structural variants
Breakpoint track (BP)
The breakpoint track (figure 2.4) contains a row for each called breakpoint with the following information
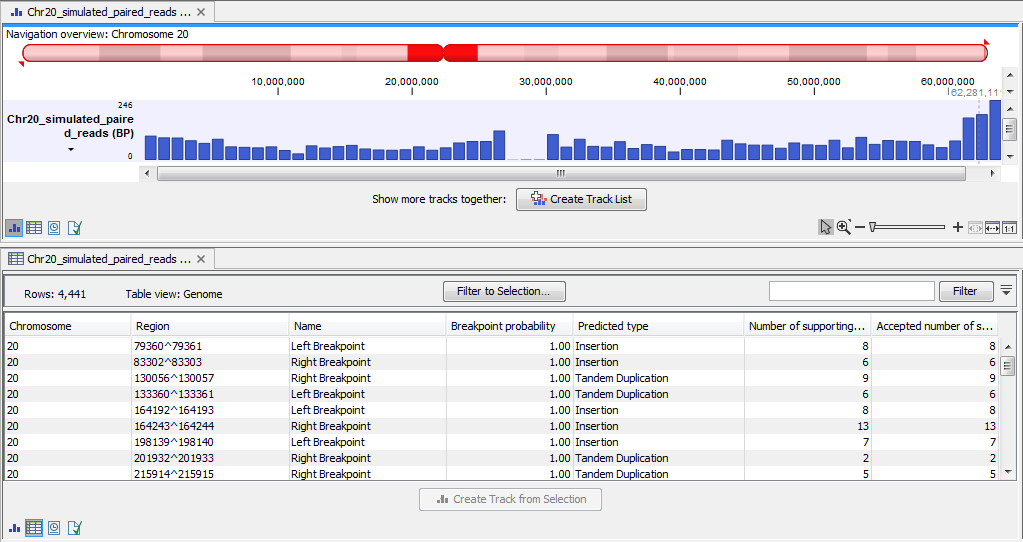
Figure 2.4: The Advanced Structural Variant Detection (beta) breakpoint track.
- Chromosome Chromosome on which the breakpoint is located.
- Region Location on the chromosome of the breakpoint.
- Name Type of the breakpoint ('left breakpoint' or 'right breakpoint').
- Breakpoint probability Estimate for how trustworthy the prediction is.
- Predicted type Whether the variant is a tandem duplication, or a deletion or insertion. Note that for tandem duplications only one duplication is reported, even in cases where a sequence appears in more than two copies in the reads.
- Number of supporting unaligned ends Number of reads at the breakpoint position with an unaligned end.
- Accepted number of supporting unaligned ends Number of reads at the breakpoint position with an unaligned end, after spurious unaligned ends have been filtered away.
Variant track (SV) and Variant as annotation track (SV annotations)
The variant tracks (figure 2.5) contain a row for each of the called structural variations. In the case of the annotated track, the structural variations are stored as annotations: large deletions may be easier to visualize in this format.
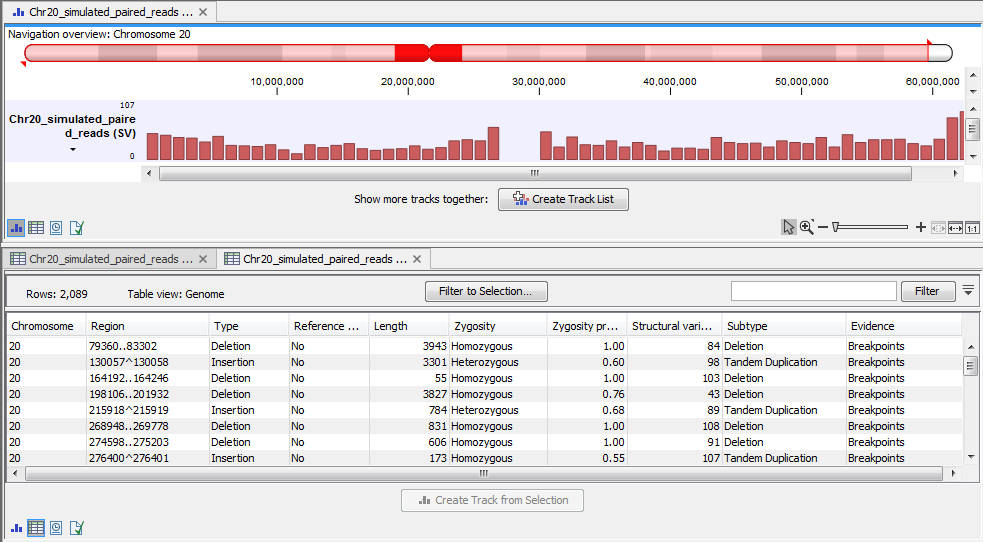
Figure 2.5: The Advanced Structural Variant Detection (beta) variant track.
In addition to the regular variant tracks information, the variant table contains the following information:
- Zygosity probability Estimate for how likely the zygosity prediction is.
- Structural variation score Measure of how likely this structural variant is.
- Subtype Available for the SV track only. This is a more specific categorization of the structural variant type: either Insertion, Deletion, Tandem Duplication, CNV Loss, or CNV Gain. Note that for Tandem Duplications only one duplication is reported, even in cases where a sequence appears in more than two copies in the reads.
- Evidence May be either Single Breakpoint, Paired Breakpoints, CNV + Breakpoint (i.e., based on coverage information and a single breakpoint), or Broken pairs (in which case the sequence may not be accurate). The broken pairs option is special since it is based on assembly of broken read pairs, where one of the reads in a pair maps at a different location in the genome. This allows for detection of insertions of Alu elements for example. Note that the sequence here may not be accurate since it is based on a homologous region of the genome.