RNA-seq
Based on an annotated reference genome, CLC Genomics Workbench supports RNA-Seq Analysis by mapping next-generation sequencing reads and distributing and counting the reads across genes and transcripts. Subsequently, the results can be used for expression analysis. The tools from the RNA-Seq folder automatically account for differences due to sequencing depth, removing the need to normalize input data. The statistical analysis and visualization tools of the RNA-Seq folder make extensive use of the metadata system.
Metadata for RNA-Seq
The statistical analysis and visualization tools of the RNA-Seq folder make extensive use of the metadata system. For example, metadata are required when defining the experimental design in the Differential Expression for RNA-Seq tool, and can be used to add extra layers of insight in the Create Heat Map and PCA for RNA-Seq tools.
To get the most out of these tools we recommend that all input expression tracks have associated metadata, as shown in figure 28.1. For information about how to use and setup metadata, please see Metadata.
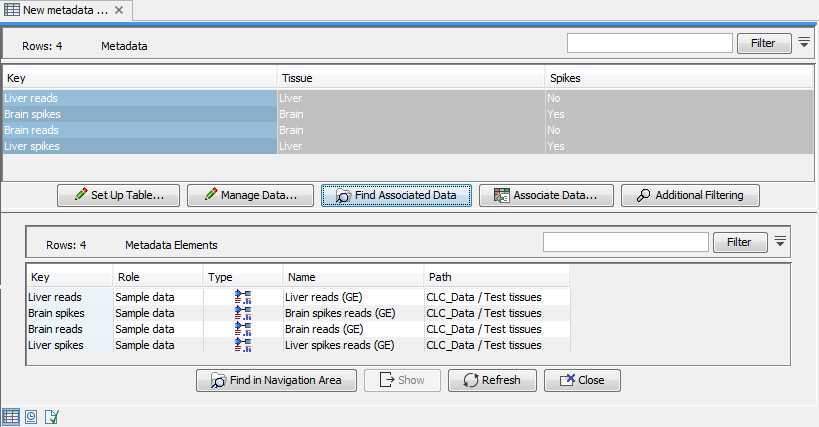
Figure 28.1: An example of expression tracks with associated metadata.
Subsections
- RNA-seq normalization
- RNA-Seq Analysis
- Create Combined RNA-Seq Report
- PCA for RNA-Seq
- Differential Expression
- Create Heat Map for RNA-Seq
- Create Expression Browser
- Create Venn Diagram for RNA-Seq
- Gene Set Test