RNA-Seq analysis
The following describes the overall process of the RNA-Seq analysis when using an annotated eukaryote genome. See Specifying reads, reference genome and mapping settings for more information on other types of reference data.
The RNA-Seq analysis is done in several steps: First, all annotated transcripts are extracted (using an mRNA track). If there are several annotated splice variants, they are all extracted.
An example is shown in figure 29.2.
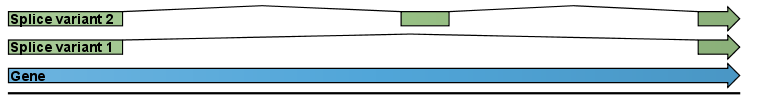
Figure 29.2: A simple gene with three exons and two splice variants.
This is a simple gene with three exons and two splice variants. The transcripts are extracted as shown in figure 29.3.
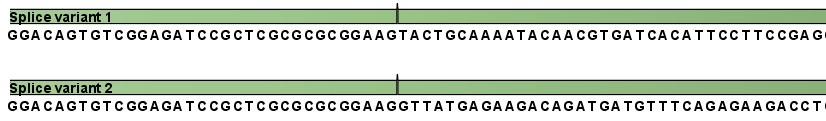
Figure 29.3: All the exon-exon junctions are joined in the extracted transcript.
Next, the reads are mapped against all the transcripts plus to the whole genome. For more information about the read mapper, see Map Reads to Reference.
From this mapping, the reads are categorized and assigned to the genes using the EM estimation algorithm , and expression values for each gene and each transcript are calculated.
Subsections
- Specifying reads and reference
- Defining mapping options for RNA-Seq
- The EM estimation algorithm
- Calculating expression values from RNA-Seq
- Specifying RNA-Seq outputs
- Expression tracks
- Reads track
- RNA-Seq report
- Gene fusion reporting