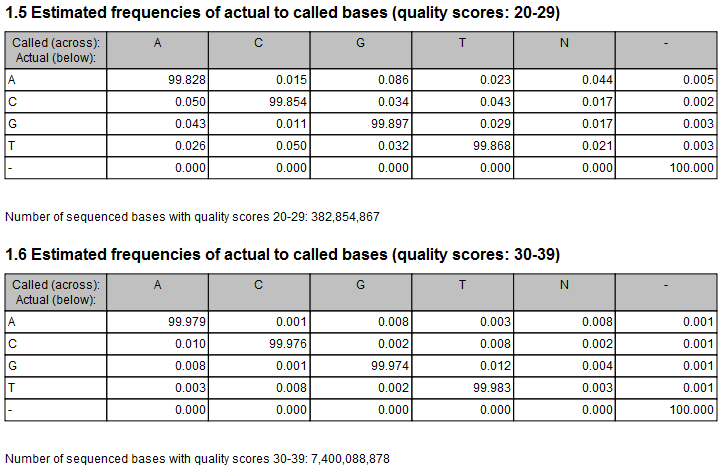
Figure 27.19: Example of estimated error rates. The figure shows average estimated error rates across bases in the given quality score intervals (20-29 and 30-39, respectively). Higher error rates are estimated for bases with lower quality scores.
Figure 27.19: Example of estimated error rates. The figure shows average estimated error rates across bases in the given quality score intervals (20-29 and 30-39, respectively). Higher error rates are estimated for bases with lower quality scores.
An example of error rates estimated from a whole exome sequencing Illumina data set is shown in figure 27.19. As expected, the estimated error rates (that is, the off-diagonal elements in the matrices in the figure) are higher for the lower quality nucleotides than for higher. Note that, although the matrices in the figure show error rates of bases within ranges of quality scores, a separate matrix is estimated for each quality score in the error model estimation.