How side chains are modeled
Amino acid side chains tend to assume one of a discrete number of "rotamer" conformations. The rotamers used in CLC Genomics Workbench have been calculated from a non-redundant set of high-resolution crystal structures.
Side chains are modeled using a heat bath Monte Carlo simulated annealing algorithm,
similar to the OPUS-Rota method [Lu et al., 2008]. The algorithm consists of approximately 100 cycles of simulation. In a single cycle, rotamers are selected for each side chain with a probability according to their energy. As the simulation proceeds, the selection increasingly favors the rotamers with the lowest energy, and the algorithm converges.
A local minimization of the modeled side chains is then carried out, to reduce unfavorable interactions with the surroundings.
Calculating the energy of a side chain rotamer
The total energy is composed of several terms:
- Statistical potential: This score accounts for interactions between the given side chain and the local backbone, and is estimated from a database of high-resolution crystal structures. It depends only on the rotamer and the local backbone dihedral angles
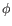 and
and 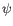 .
.
- Atom interaction potential: This score is used to evaluate the interaction between a given side chain atom and its surroundings. It is the same score that is used to evaluate protein-ligand interactions for docking and ligand optimization in CLC Drug Discovery Workbench.
- Disulfide potential: Only applies to cysteines. It follows the form used in the RASP program [Miao et al., 2011] and serves to allow disulfide bridges between cysteine residues. It penalizes deviations from ideal disulfide geometry. A distance filter is applied to determine if the disulfide potential should be used, and when it is applied the atom interaction potential between the two sulfur atoms is turned off. Note that disulfide bridges are not formed between separate chains.
|
Note: The atom interaction potential considers interactions within the modeled protein chain as well as with all other molecules in the downloaded PDB file (except water).
|
Local minimization of side chain
After applying a side chain rotamer from the library to the backbone, a local minimization may be carried out for rotations around single bonds in the side chain.
The potential to minimize with respect to bond rotation is composed of the following terms:
- Atom interaction potential: Same as for calculating the energy of a rotamer.
- Disulfide potential: Same as for calculating the energy of a rotamer.
- Harmonic potential: This penalizes small deviations from ideal rotamers according to a harmonic potential. This is motivated by the concept of a rotamer representing a minimum energy state for a residue without external interactions.