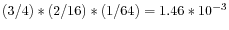Procedure for inspecting breakpoint signatures and inferring Structural Variants
The procedure for inspecting the created LBs and RBs and inferring structural variants from them works as follows:
- Calculate unaligned end consensus: For each breakpoint, we calculate the consensus of the unaligned ends. We do this by simple alignment without gaps. Having created the consensus, we exclude the unaligned ends which differ by more than 20% from the consensus, and recalculate the consensus. This prevents 'spuriously' unaligned ends that extend longer than other unaligned ends from impacting the tail of the consensus unaligned end. We say that a consensus unaligned end 'exists' if it is > 4 nucleotides long. For some breakpoints no unaligned end consensus will exist - either because it is too short, or because there are no reads left to calculate the consensus from (e.g. there is no consensus unaligned end for a breakpoint with two reads with completely different unaligned ends, even when they are longer than 4 nucleotides).
- 'Self-map' the unaligned end consensus: For each breakpoint, if the consensus unaligned end 'exists' and is long enough to be meaningfully mapped (we use 14nt or longer), we map the consensus unaligned end to the reference surrounding the breakpoint (we search in a window extending from 100 bp upstream to 100 bp downstream of the breakpoint). If it maps, a note is made that it is self-mapped.
- 'Link' breakpoint signatures: This step serves to determine which breakpoint signatures originate from the same structural variant. To do this, we go through all breakpoints and create a 'group' of breakpoints consisting of the ones with which the breakpoint is 'linked'. There are two ways in which breakpoints can be linked: either as 'Close' breakpoints or as 'Cross-mapped' breakpoints. These linkings are defined as follows:
- 'Close' breakpoints: unaligned end breakpoints are linked as 'close breakpoints' if they are either less than 5 nucleotides or the "length of the unaligned end consensus" nucleotides apart.
- 'Cross-mapped' breakpoints: two unaligned end breakpoints, X and Y, are cross mapped if all of the following apply:
- They are NOT 'Close' breakpoints.
- One is an LB and the other an RB.
- The unaligned end of X maps to the reference sequence around Y (we search in a window extending from 100 bp upstream to 100 bp downstream of the breakpoint Y)
- The unaligned end of Y maps to the reference sequence around X (we search in a window extending from 100 bp upstream to 100 bp downstream of the breakpoint X)
- the mappings above are on the same strand.
Note that 'Cross-mapped' breakpoints are also created if there are multiple hits. E.g.: If we have breakpoints A, B, C, D, E and F, and if A maps B,C and D, and C maps to A, E and F, A and C will be cross mapped.
- 'Process breakpoint groups': Next we go through all the 'groups' of breakpoints created in the linking step above. Depending on the number (whether there is a single, two or more than two) and on the types of breakpoint signatures in a group, we do the following:
- One breakpoint:
- If the breakpoint is 'Self-mapped', create an 'Insertion (mapped)' or a 'Deletion (mapped)' signature. Whether an insertion or deletion is made, depends upon how the unaligned end self-maps to the reference.
- If the breakpoint is not self mapped, do not create any signature.
- Two 'close' breakpoints:
- If the two breakpoints are NOT an LB and an RB: de-group the breakpoints and process the breakpoints as two single breakpoints.
- If the two breakpoints ARE an LB and an RB: extend the two unaligned end sequences to also include 50 bp of the reference sequence respectively upstream and downstream of the unaligned end starting points. Align the resulting sequences (using the "Merge Overlapping Paired Read" aligner). Consider the consensus of this alignment:
- if no consensus could be constructed: create an 'Insertion (close breakpoints)' signature.
- if a consensus could be constructed: align the consensus to the reference sequence, and create a structural variant signature, depending on how the resulting alignment looks. There are three cases:
- The alignment contains gaps in the reference: we create an 'Insertion (paired breakpoints)' signature.
- The alignment contains gaps in the consensus: we create a 'Deletion (close breakpoints)' signature.
- The alignment contains mismatches: we create a 'Replacement (paired breakpoints)'
If the 'Replacement" has the same length in the reference and consensus and the length is significant. If the replacements is a reverse complement of itself: we create 'Inversion (paired breakpoints)' signature.
- if the consensus is smaller than the reference: create a 'Deletion (close breakpoints)' signature.
- if the consensus is larger than the reference: create an 'Insertion (paired breakpoints)' signature.
- if a part of the consensus doesn't match the reference and the consensus is a perfect and 'significant' inversion of the reference: create an 'Inversion (paired breakpoints)' signature.
- if a part of the consensus doesn't match the reference and the consensus is NOT a perfect and 'significant' inversion of the reference: create a 'Replacement (paired breakpoints)' signature.
(For 'significance' we use the length of the inverted sequence 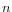 , and estimate the probability that the replaced bases occur in exactly the 'inversion' order as
, and estimate the probability that the replaced bases occur in exactly the 'inversion' order as 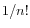 If this value is smaller than the user-specified p-value cut-off, the inversion is said to be 'significant'. The default p-value of 0.0001 requires the length of the inverted sequence to be at least 8, in order to deem it 'significant' and thus call an 'inversion' rather than a 'replacement').
If this value is smaller than the user-specified p-value cut-off, the inversion is said to be 'significant'. The default p-value of 0.0001 requires the length of the inverted sequence to be at least 8, in order to deem it 'significant' and thus call an 'inversion' rather than a 'replacement').
- Two 'cross mapped' breakpoints:
- If the RB lies left of the LB we create a 'Deletion (cross-mapped)' signature.
- If the LB lies left of the RB we create a 'Tandem duplication' signature.
- If the mappings are found on the reverse complement strand we create an 'Inversion' signature.
- One close and one cross mapped breakpoint: Not possible as we do not attempt to cross map breakpoints to close breakpoints.
- More than two breakpoints: Groups with more than two breakpoints represent structural variants with more complicated unaligned end breakpoint signatures. Each group is processed as follows:
- If there are two or more 'cross mapped' breakpoints in the group, we take the 'cross mapped' pair that has the shortest distance between them and process them as described under 'Two cross mapped breakpoints'. Others are ignored. This will create a 'Deletion (cross mapped)', 'Tandem duplication' or 'Inversion' signature. Add this to the group.
- All pairs of 'close' breakpoints in the group -- EXCEPT those (if any) that were also the two chosen 'cross' mapped breakpoints in above -- are processed as 'Two close breakpoints'. This will create deletion, insertion or replacement signatures. Add these to the group.
- If the group, after the above processing, has a deletion and an insertion signature: create a 'Translocation' signature.
- For remaining groups: create a 'Complex variant' signature.
- Create structural variant features: For each structural variant signature present after step 4 - except those that extend over too large a region! - , we create a structural variant feature. For those extending over too large a region, visualization is challenging and we instead add multiple features - one for each 'end' of the variant. To allow the user to see which of these 'split features' belong together, we give features that belong to the same structural variant a common 'split group' identifier.
- Add repeat information: Augment the predicted structural variants with repeat information: For structural variants for which we have identified the variant sequence (there are some, e.g. larger insertions, for which this is not possible) we attempt to identify if the variant sequence contains (perfect) repeats. We do this by searching the region around the structural variant for perfect repeat sequences. The region searched is 3 times the length of variant around the insertion/deletion point.
- Add information regarding 'zygosity' ('Variant ratio'): Augment the predicted breakpoints and structural variants with information related to zygosity: For all unaligned end breakpoints we examine all the reads that cover the breakpoint. We count the number of reads that are mapped across this position and have either an unaligned end or insertions or deletions in their mappings. We call these the 'Non-perfect mapped reads'. The remaining mapped reads that cover the position (that is, those that are mapped in their entire length, and for which there are no insertions or deletions in the alignment) are called 'Perfect mapped reads'. For breakpoints we report the fraction of the 'Non-perfect mapped' reads among all mapped reads (that is: 'Non-perfect mapped'/('Non-perfect mapped'+'Perfect mapped')). For a Structural variant that is generated from more breakpoints, we sum the number of reads for the individual breakpoints and report the fraction of these in the 'Variant ratio' column. This fraction is intended to give some idea of the zygosity of the breakpoint or structural variant. The closer the value to 1, the higher the likelihood that the variant is homozygous. However, as the 'Non-perfect mapped reads' include all reads with any unaligned end or any insertion or deletion, and not only the ones that are in perfect accordance with the breakpoint or structural variant called, it is NOT a perfect indicator of zygosity. It does, however, often give a good indication.
- Add information regarding the breakpoints used to construct the structural variant: For each structural variant a number of values related to the unaligned end breakpoints used to construct the predicted variant are recorded. These are provided in order to let the user assess the degree of evidence supporting the predicted structural variant. The values are: The number of breakpoints used to construct the variant, their positions, the mapping scores and sequence complexities of the unaligned ends, as well as the numbers of reads supporting them. The mapping scores are the similarity values between the unaligned end at the region of the reference to which it was mapped. These are values between 0 and 1, and the closer to 1, the better the match and thus the more reliable the inferred variant. The sequence complexity of an unaligned end is calculated as the product of the vocabulary-usage measures for word sizes up to five, and when multiple breakpoints are used to construct a structural variant, the complexity is calculated as the product of the individual complexities of the breakpoints 26.2.
The algorithm has been found to predict some (typically longer and/or of type "complex") structural variants from unaligned end breakpoints with large support in terms of unaligned end lengths and supporting reads, but with low-complexity sequences. These are likely to be false positives, and the complexity values are provided to allow the user to be alerted of these. Finally, the number of reads supporting a predicted structural variant is defined to be the sum of the numbers of reads supporting the breakpoints used to construct the structural variant.
Footnotes
- ... breakpoints26.2
- The vocabulary usage for an oligomer of a given size, k, can be defined as the ratio of the actual vocabulary size of a given sequence to the maximal possible vocabulary size for a sequence of that length. For example, the vocabulary usages U1, U2 and U3, for the oligomer AAA are 1/4, 1/16 and 1/64, and for the sequence GAC they are 3/4, 2/16 and 1/64. The complexity of the AAA oligomer is thus
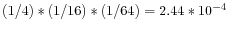 and of the oligomer GAC
and of the oligomer GAC