You can view and edit the orientation of the reads after they have been imported by opening the read list in the Element information view (![]() ) as shown in figure 6.12.
) as shown in figure 6.12.
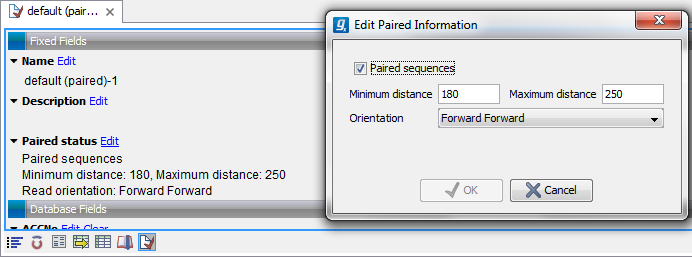
Figure 6.12: The paired orientation and distance.
In the Paired status part, you can specify whether the CLC Genomics Workbench should treat the data as paired data, what the orientation is and what the preferred distance is. The orientation and preferred distance is specified during import and can be changed in this view.
Note that the paired distance measure that is used throughout the CLC Genomics Workbench is always including the full read sequence. For paired-end libraries it means from the beginning of the forward read to the beginning of the reverse read.