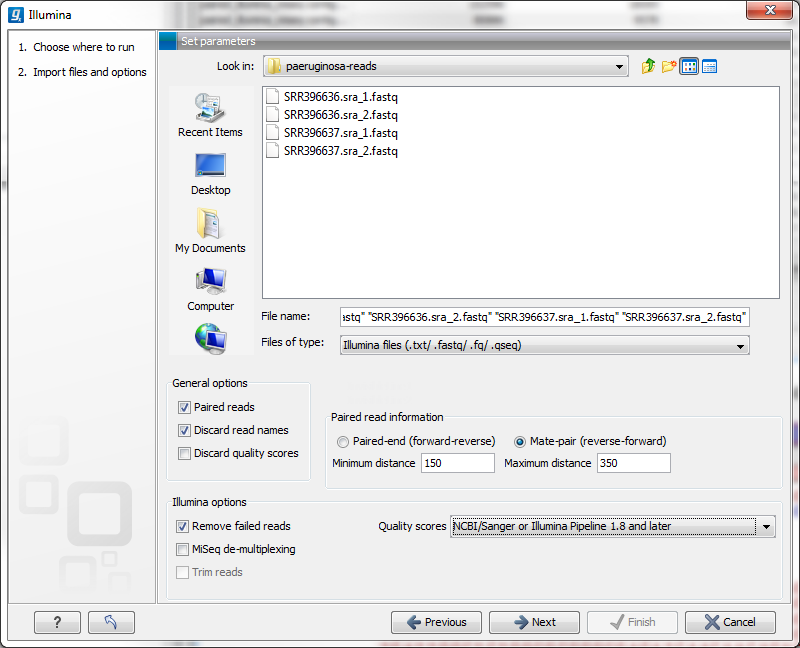
Figure 6.5: Importing data from Illumina systems.
Figure 6.5: Importing data from Illumina systems.
The file formats accepted are:
Note that there is information inside qseq and fastq files specifying whether a read has passed a quality filter or not. If you check Remove failed reads these reads will be ignored during import. For qseq files there is a flag at the end of each read with values 0 (failed) or 1 (passed). In this example, the read is marked as failed and if Remove failed reads is checked, the read is removed.
M10 68 1 1 28680 29475 0 1 CATGGCCGTACAGGAAACACACATCATAGCATCACACGA BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB 0
For fastq files, part of the header information for the quality score has a flag where Y means failed and N means passed. In this example, the read has not passed the quality filter:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
Note! In the Illumina pipeline 1.5-1.7, the letter B in the quality score has a special meaning. 'B' is used as a trim clipping. This means that when selecting Illumina pipeline 1.5-1.7, the reads are automatically trimmed when a B is encountered in the input file. This will happen also if you choose to discard quality scores during import.
If you import paired data and one read in a pair is removed during import, the remaining mate will be saved in a separate sequence list with single reads.
For all formats, compressed data in gzip format is also supported (.gz).
The General options to the left are:
When loading files containing paired data, the CLC Genomics Workbench sorts the files selected according to rules based on the file naming scheme:
_R1_ are assumed to contain the first sequences of the pairs, and those with _R2_ in the name are assumed to contain the second sequence of the pairs.
In the simplest case, the files are typically named as shown in figure 6.5. In this case, the data is paired end, and the file containing the forward reads is called s_1_1_sequence.txt and the file containing reverse reads is called s_1_2_sequence.txt. Other common filenames for paired data, like _1_sequence.txt, _1_qseq.txt, _2_sequence.txt or _2_qseq.txt will be sorted alphanumerically. In such cases, files containing the final _1 should contain the first reads of a pair, and those containing the final _2 should contain the second reads of a pair.
For files from CASAVA1.8, files with base names like these: ID_R1_001, ID_R1_002, ID_R2_001, ID_R2_002 would be sorted in this order:
The data in files ID_R1_001 and ID_R2_001 would be loaded as a pair, and ID_R1_002, ID_R2_002 would be loaded as a pair.
Within each file, the first read of a pair will have a 1 somewhere in the information line. In most cases, this will be a /1 at the end of the read name. In some cases though (e.g. CASAVA1.8), there will be a 1 elsewhere in the information line for each sequence. Similarly, the second read of a pair will have a 2 somewhere in the information line - either a /2 at the end of the read name, or a 2 elsewhere in the information line.
If you do not choose to discard your read names on import (see next parameter setting), you can quickly check that your paired data has imported in the pairs you expect by looking at the first few sequence names in your imported paired data object. The first two sequences should have the same name, except for a 1 or a 2 somewhere in the read name line.
Paired-end and mate-pair data are handled the same way with regards to sorting on filenames. Their data structure is the same the same once imported into the Workbench. The only difference is that the expected orientation of the reads: reverse-forward in the case of mate pairs, and forward-reverse in the case of paired end data. Read more about handling paired data.
Click Next to adjust how to handle the results. We recommend choosing Save in order to save the results directly to a folder, since you probably want to save anyway before proceeding with your analysis. There is an option to put the import data into a separate folder. This can be handy for better organizing subsequent analysis results and for batch processing.