Figure 28.12: A bubble caused by a heteroygous SNP or a sequencing error.
![]()
Figure 28.12: A bubble caused by a heteroygous SNP or a sequencing error.
In this simple case the assembler will collapse the bubble and use the variant that has the highest count of words. For a diploid genome with a heterozygous variant, there will a fifty-fifty distribution of reads on the two variants, and this means that the choice of one allele over the other will be arbitrary. If heterozygous variants are important, they can be identified after the assembly by mapping the reads back to the contig sequences and performing standard variant calling. For random sequencing errors, it is more straightforward; given a reasonable level of coverage, the erroneous variant will be suppressed.
Figure 28.13 shows an example of a data set where the reads have systematic errors. Some reads include five As and others have six. This is a typical example of the homopolymer errors seen with the 454 and Ion Torrent platforms.

Figure 28.13: Reads with systematic errors.
When these reads are assembled, this site will give rise to a bubble in the graph. This is not a problem in itself, but if there are several of these sites close together, the two paths in the graph will not be able to merge between each site. This happens when the distance between the sites is smaller than the word size used (see figure 28.14).
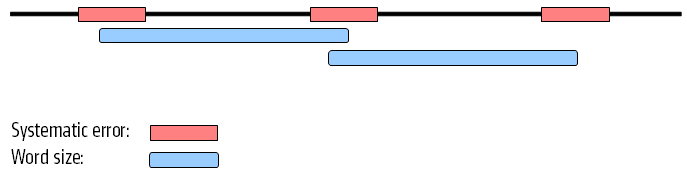
Figure 28.14: Several sites of errors that are close together compared to the word size.
In this case, the bubble will be very large because there are no complete words in the regions between the homopolymer sites, and the graph will look like figure 28.15.
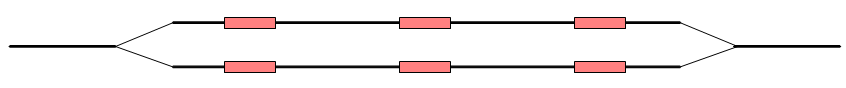
Figure 28.15: The bubble in the graph gets very large.
If the bubble is too large, the assembler will have to break it into several separate contigs instead of producing one single contig.
The maximum size of bubbles that the assembler should try to resolve can be set by the user. In the case from figure 28.15, a bubble size spanning the three error sites will mean that the bubble will be resolved (see figure 28.16).
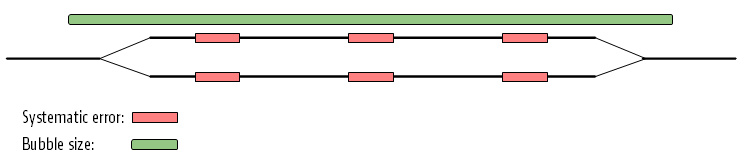
Figure 28.16: The bubble size needs to be set high enough to encompass the three sites.
While the default bubble size is often fine when working with short, high quality reads, considering the bubble size can be especially important for reads generated by sequencing platforms yielding long reads with either systematic errors or a high error rate. In such cases, a higher bubble size could be recommended. For example, as a starting point, one could try half the length of the average read in the sequence set. In many situations, you might then wish to investigate the effect of decreasing the bubble size. If you also investigate the effect of increasing tHe bubble size, please keep in mind that increasing this too much can potentially lead to misassemblies with some data sets.