Join alignments
CLC Genomics Workbench can join several alignments into one. This feature can for example be used to construct "supergenes" for phylogenetic inference by joining alignments of several disjoint genes into one spliced alignment. Note, that when alignments are joined, all their annotations are carried over to the new spliced alignment.
Alignments can be joined by:
select alignments to join | Toolbox in the Menu
Bar | Classical Sequence Analysis (![]() ) | Alignments and Trees (
) | Alignments and Trees (![]() )|
Join Alignments (
)|
Join Alignments (![]() )
)
or select alignments to join | right-click
either selected alignment | Toolbox | Classical Sequence Analysis (![]() ) | Alignments and Trees (
) | Alignments and Trees (![]() )| Join Alignments
(
)| Join Alignments
(![]() )
)
This opens the dialog shown in figure 3.7.
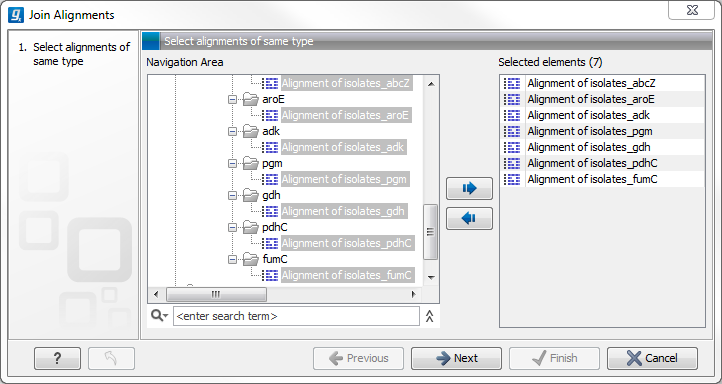
Figure 3.7: Selecting two alignments to be joined.
If you have selected some alignments before choosing the Toolbox action, they are now listed in the Selected Elements window of the dialog. Use the arrows to add or remove alignments from the selected elements. In this example seven alignments are selected. Each alignment represents one gene that have been sequenced from five different bacterial isolates from the genus Nisseria. Clicking Next opens the dialog shown in figure 3.8.
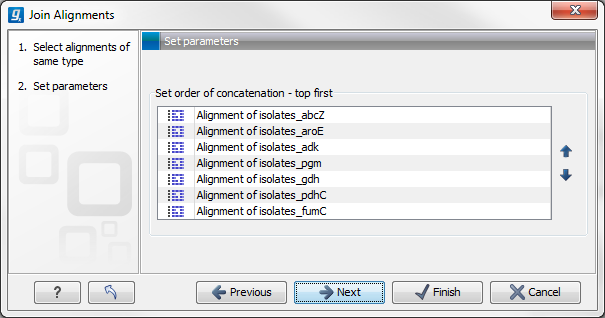
Figure 3.8: Selecting order of concatenation.
To adjust the order of concatenation, click the name of one of the alignments, and move it up or down using the arrow buttons.
The result is seen in the lower part of figure 3.9.
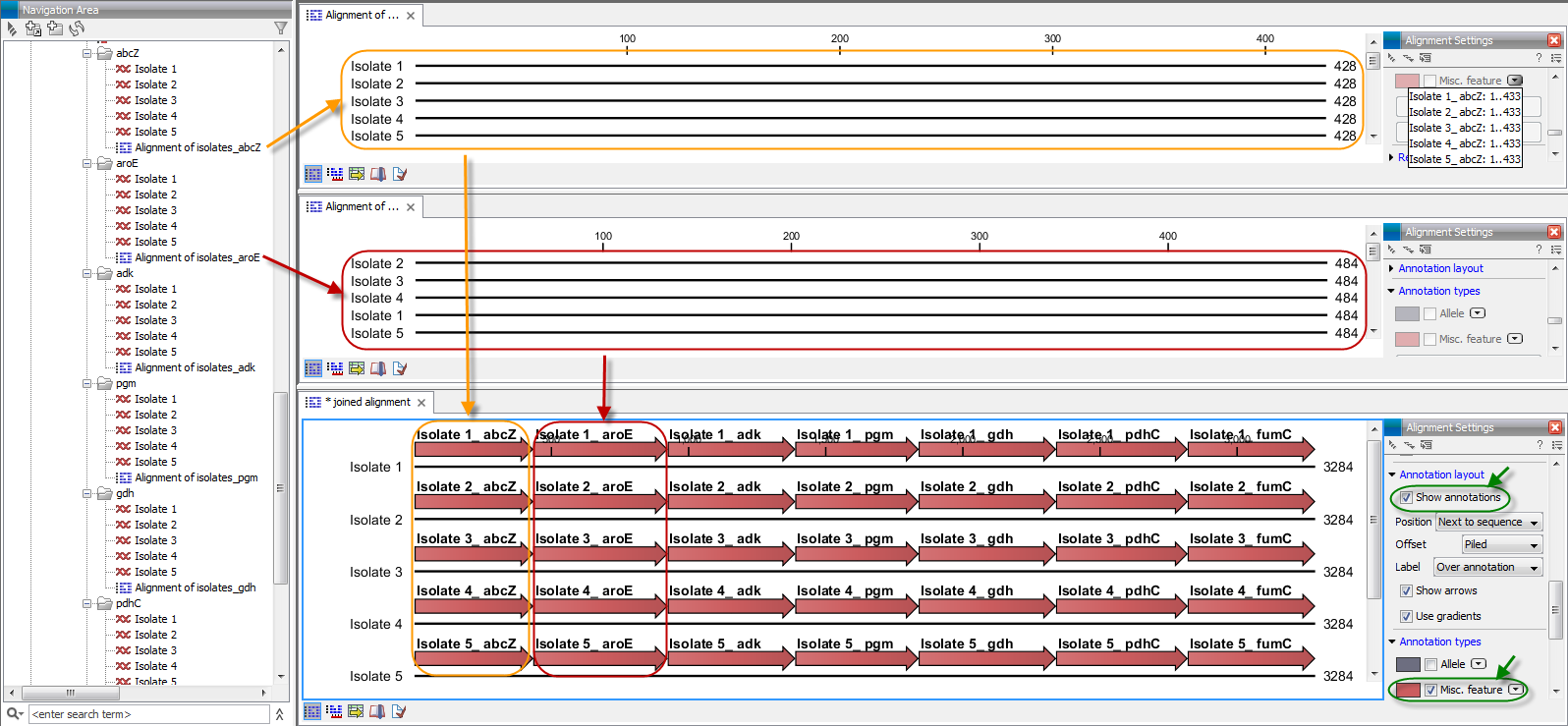
Figure 3.9: The upper part of the figure shows two of the seven alignments for the genes "abcZ" and "aroE" respectively. Each alignment consists of sequences from one gene from five different isolates. The lower part of the figure shows the result of "Join Alignments". Seven genes have been joined to an artificial gene fusion, which can be useful for construction of phylogenetic trees in cases where only fractions of a genome is available. Joining of the alignments results in one row for each isolate consisting of seven fused genes. Each fused gene sequence corresponds to the number of uniquely named sequences in the joined alignments.
Subsections