Direction and position filters
Many sequencing protocols are prone to various types of amplification induced biases and errors. The 'Read direction' and 'Read position' filters are aimed at providing means for weeding out variants that are likely to originate from such biases.
- Read direction filter: The read direction filter removes variants that are almost exclusively present in either forward or reverse reads. For many sequencing protocols such variants are most likely to be the result of amplification induced errors. Note, however, that the filter is NOT suitable for amplicon data, as for this you will not expect coverage of both forward and reverse reads. The filter has a single parameter:
- Direction frequency: Variants that are not supported by at least this frequency of reads from each direction are removed.
- Relative read direction filter: The relative read direction filter attempts to do the same thing as the 'Read direction filter', but does this in a statistical, rather than absolute, sense: it tests whether the distribution among forward and reverse reads of the variant carrying reads is different from that of the total set of reads covering the site. The statistical, rather than absolute, approach makes the filter less stringent. The filter has one parameter:
- Significance: Variants whose read direction distribution is significantly different from the expected with a test at this level are removed. The lower you set the significance cut-off, the fewer variants will be filtered out.
- Read position filter: The read position filter is a filter that attempts to remove systematic errors in a similar fashion as the 'Read direction filter', but that is also suitable for hybridization-based data. It removes variants that are located differently in the reads carrying it than would be expected given the general location of the reads covering the variant site. This is done by categorizing each sequenced nucleotide (or gap) according to the mapping direction of the read and also where in the read the nucleotide is found; each read is divided in five parts along its length and the part number of the nucleotide is recorded. This gives a total of ten categories for each sequenced nucleotide and a given site will have a distribution between these ten categories for the reads covering the site. If a variant is present in the site, you would expect the variant nucleotides to follow the same distribution. The read position filter carries out a test for whether the read position distribution of the variant carrying reads is different from that of the total set of reads covering the site. The filter has one parameter:
- Significance: Variants whose read position distribution is significantly different from the expected with a test at this level, are removed. The lower you set the significance cut-off, the fewer variants will be filtered out.
Figure 25.39 shows an example of a variant that is removed by the 'Read direction' filter. To see the direction of the reads, you must adjust the viewer settings in the 'Reads track' side panel to 'Disconnect paired reads'. Note that variant calling was done ignoring non-specific matches and broken pair reads, so only the 16 intact forward paired reads (the green reads) are considered. In this example there was no intact reverse reads.
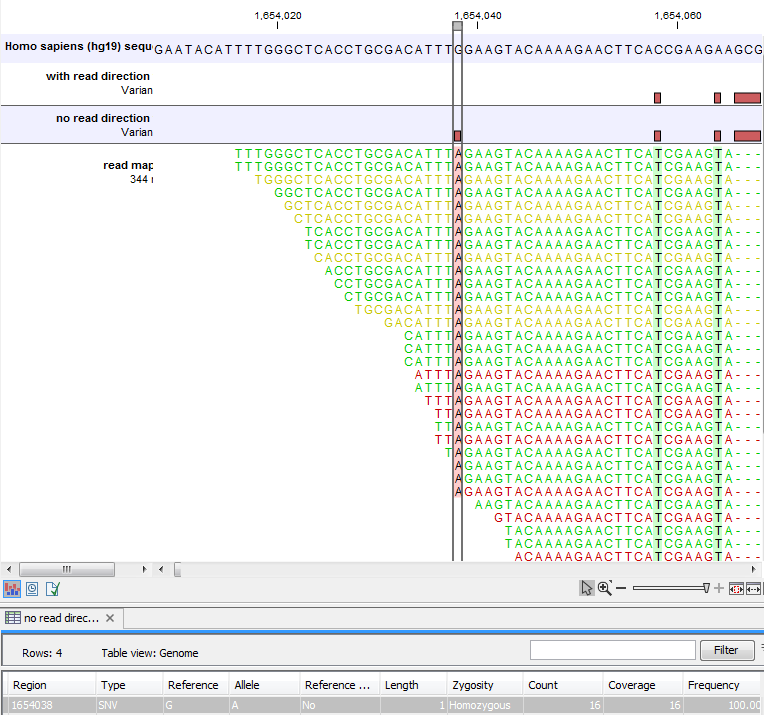
Figure 25.39: Example of a variant that is removed by the 'Read direction' filter.
Figure 25.40 shows an example of a variant that is removed by the 'Read position' filter, but not by the 'Read direction' filter. This variant is only seen in a set of reads having a similar start position, while reads that start in a different location do not contain this variant (e.g., none of the reads that start after position 186,641,600 carry the variant). This could indicate the incorporation of an incorrect base during the library preparation process rather than a true biological variant. The purpose of the 'Read position' filter is to reduce the presence of these types of variants. As with all noise filters, the more stringent the setting, the more likely you are to remove false positives and enrich your result for true positive variant calls but comes with the risk of filtering out true positives as well.
Understanding the type of false positive this filter is intended to remove will help you to determine what makes sense for your data set. For example, if your sequencing data did not include a PCR step or hybrid capture step, you may wish to use more lax settings for this filter (or not use it at all).
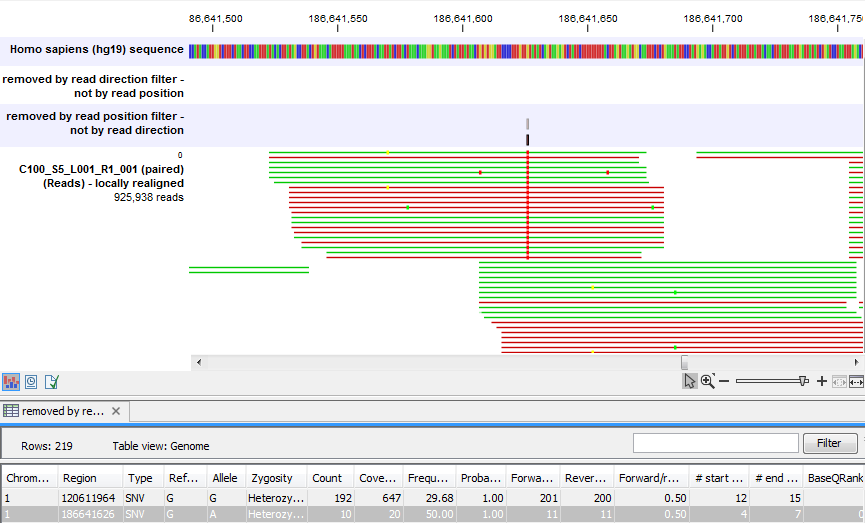
Figure 25.40: A variant that is filtered out by the Read position filter but not by the Read direction filter.