Screen ligands
In virtual screenings, a large general library of drug-like compounds are docked one by one to the target protein. The top-scoring ligands are expected to present with ligands that will either be candidates for strong binders in their own right, or give insight into molecule elements that are particular important for strong binding to the particular binding pocket.
The Screen Ligands tool use the same docking algorithms (see The docking algorithms) as the Dock Ligands tool (Ligand docking using the Dock Ligands tool). The Screen Ligands wizard (figure 9.61) thus needs the same input and the parameters in step 2 are the same, and have the same meaning, as for the Dock Ligands tool (Ligand docking using the Dock Ligands tool), with a few exceptions listed below.
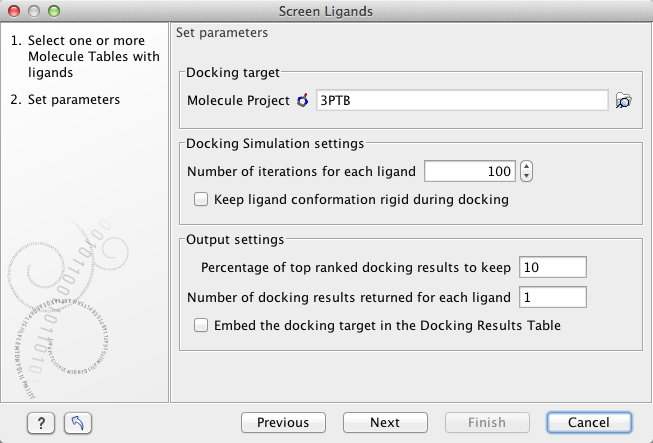
Figure 9.61: Parameter settings for virtual screening.
Percentage of top ranked docking results to keep. A virtual screening can involve Molecule Table(s) with a very large number of compounds (> 100,000). The Docking Results Table will in that case take up a considerable amount of disk space - equivalent to the input table. This can also make the results table slow to work with when e.g. sorting the entries based on a particular column. In general, it will only be the top ranked ligands that are interesting to look at, and this option therefore discards all but the top ranked docking results. When the parameter is set to 10, only the 10 % best scored ligands will be included in the results table.
The option to calculate RMSDs between input structure and results has been removed, as this does not make sense to do for a screening library.