Ligand docking using the Dock Ligands tool
To run the "Dock Ligands" tool:
Toolbox | Drug Design (![]() ) | Dock Ligands (
) | Dock Ligands (![]() )
)
The Dock Ligands tool takes a Molecule Table containing small molecules (ligands), and a Molecule Project containing a Binding Site Setup (see Setup Binding Site) as input. For each of the small molecules in the Molecule Table, the docking simulation searches for optimal binding modes to the binding site. The optimal binding mode together with a score of the binding mode are returned in a Docking Results Table (see Docking Results Tables). The docking algorithms are described in The docking algorithms.
In the Dock Ligands wizard step 1 (figure 9.55), select the Molecule Table that holds the small molecules to dock. More than one table can be selected at the same time.
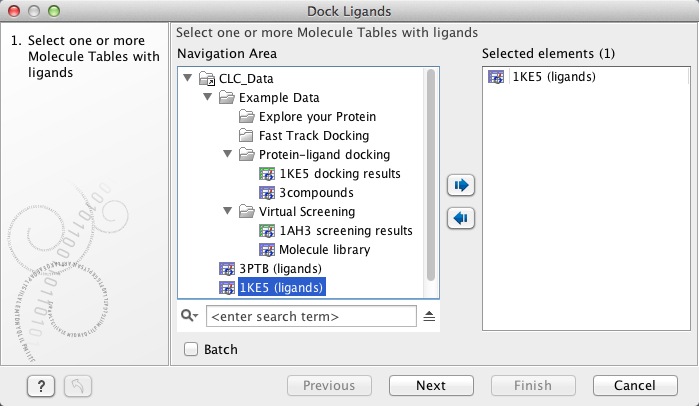
Figure 9.55: The "Dock Ligands" dialog. Select the "Molecule Table" that hold the small molecules to dock.
Click on the button labeled Next to select the Molecule Project and specify the docking parameters (figure 9.56).
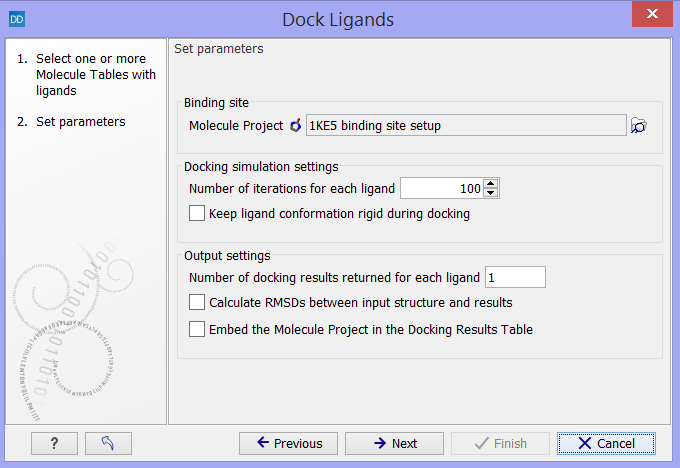
Figure 9.56: Select the "Molecule Project" with the binding site setup and specify the docking parameters.
This wizard has the following options:
- Binding site
- Molecule Project. The Molecule Project holding the Binding Site Setup to dock against should be specified.
- Docking simulation settings
- Number of iterations for each ligand. For each ligand, a number of iterations are carried out in the search for an optimal binding mode. The default value is considered to be a good balance between search completeness and cost. Increasing the number will lead to a more extensive binding mode search, at the cost of increased computational time.
- Keep ligand conformation rigid during docking. During docking, the conformation of the ligand is changed via rotation around flexible bonds. Some molecule libraries contain pre-generated conformations of the ligands, and in that case, the ligand conformations should not be altered during the docking, and this option should be checked.
- Output settings
- Number of docking results returned for each ligand. Per default, only the best scoring binding mode is returned for each ligand. As the scoring is a simple description of molecular interaction, and thus not perfect (see The docking algorithms), it can be relevant and interesting to inspect more than just the top ranked binding mode. If the number of docking results to return for each ligand is set higher than one, other optimized top ranked binding modes will be returned, together with the best ranked, in the Docking Results Table. If the returned binding modes are very similar, it is a sign that alternative binding modes with good scores could not be found in the binding mode search.
- Calculate root mean square deviations (RMSDs) between input structure and results. Usually, you will start a docking study by docking the co-crystallized ligand, if such exists, in the binding site. If the binding mode resulting from the docking is similar to the binding mode of the co-crystallized ligand, it is a sign that the binding site setup is safe to use for docking small molecules similar to the ligand co-crystallized with the protein. The root mean square deviation (RMSD) is measured between the heavy atom positions of the ligand in the co-crystallized and docked binding modes.
- Embed the Molecule Project in the Docking Results Table. Selecting this option will save a copy of the Molecule Project used as input together with the Docking Results Table output from the simulation. Then it will always be possible to inspect the resulting binding modes in the binding site setup used for the docking (see Viewing Molecule Table structures in 3D), and to see which molecules were included in the setup (see Setup Binding Site). However, it will take up more disk space, and it is therefore not set as default.
Note! The wizard will remember the user defined settings from run to run. If you wish to bring the settings back to the default settings, this can be done by clicking on the small arrow button in the lower left corner of the dialog.
Click on the button labeled Next and consider whether the Docking Results Table should open when the docking is done, or if it should be saved in the Navigation Area. A log file of the simulation process can also be made.
If you choose to save the results, the next step will allow you to specify where to save it, otherwise click on the button labeled Finish, and the docking simulation will start. You can see how it proceeds in the Processes tab in the Toolbox area.