Running the workflow in batch using Metadata
In order to run the workflow in batch, metadata must be provided to describe which DNA and RNA reads belong together. This metadata can easily be supplied from an Excel spreadsheet. A minimal spreadsheet is shown in figure 7.6. It contains the start of the sample names and a column containing pairing information.

Figure 7.6: A minimal spreadsheet for running two multimodal samples, each consisting of a DNA and an RNA part. Here, we aim to run the two S1 samples in one execution of the workflow, and to run the two S2 samples in a second execution.
Start the workflow normally, but remember to tick batch twice - once when selecting DNA reads (figure 7.7), and again when selecting RNA reads (figure 7.8).
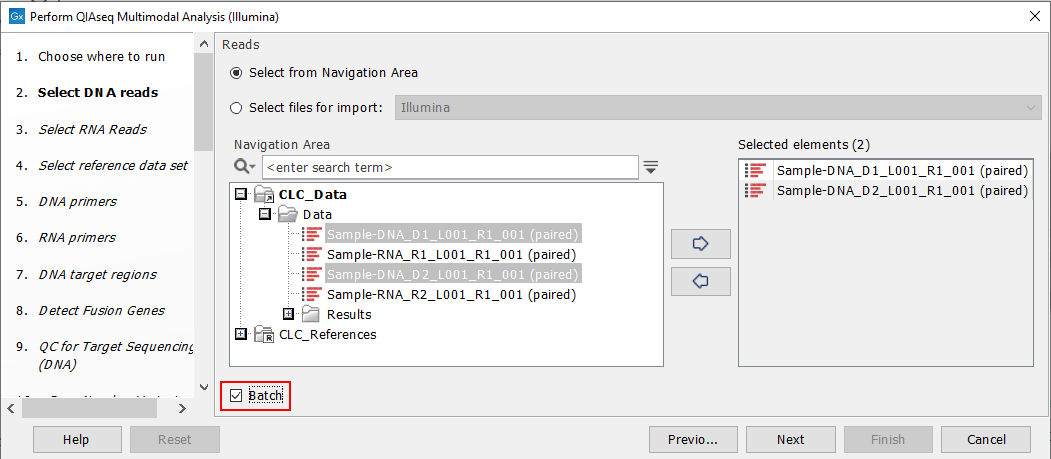
Figure 7.7: Both the input DNA samples are selected. They will be grouped into batches in a later step.
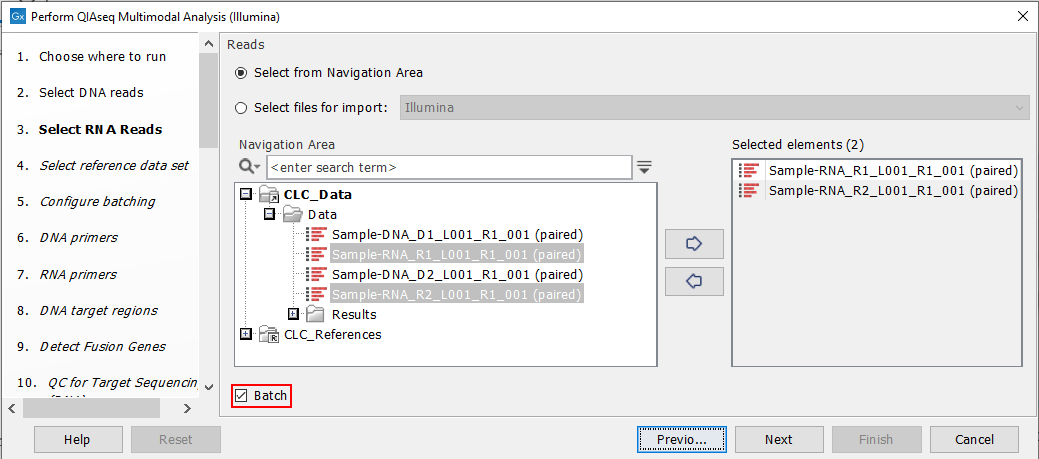
Figure 7.8: Both the input RNA samples are selected. They will be grouped into batches in a later step.
When this is done, a "Configure batching" dialog will ask for metadata. Figure 7.6 shows how to configure this:
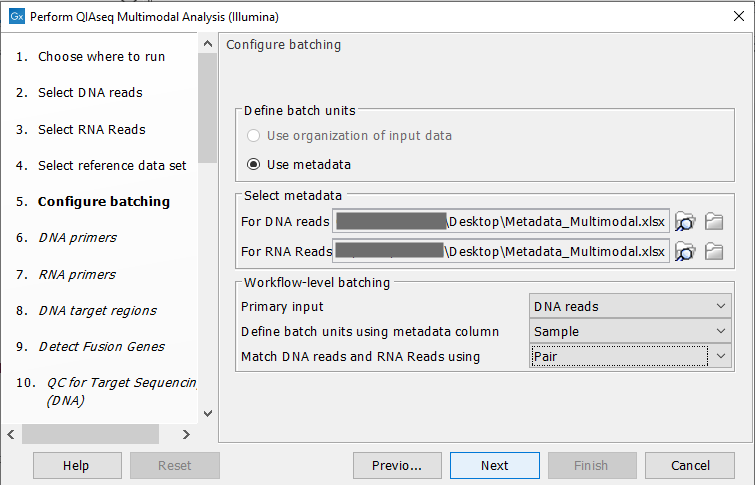
Figure 7.9: Configuration of batch units based on metadata. Each batch unit is named after the DNA sample name. DNA and RNA reads are grouped together if they share a value in the "Pair" column.
The next dialog will show you how the batching will be performed figure 7.10.
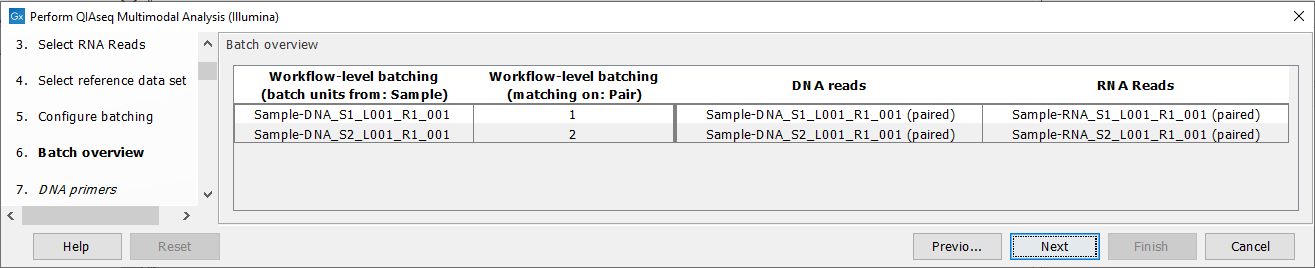
Figure 7.10: Overview of the batch units. Each batch unit is named with the DNA sample name. DNA and RNA reads are grouped together if they share a value in the "Pair" column.